LoRDEC: Frequently Asked Questions (FAQ)
Table of Contents
- 1. Abbreviations
- 2. Overview and concepts
- 2.1. Process overview
- 2.2. What is hybrid correction?
- 2.3. Why use hybrid correction?
- 2.4. Why do we need to correct long reads?
- 2.5. What is a \(k\) -mer?
- 2.6. What is a de Bruijn Graph?
- 2.7. How does LoRDEC use de Bruijn Graph?
- 2.8. Can LoRDEC perform also self-correction?
- 2.9. Can LoRDEC correct transcriptomic long reads?
- 3. Inputs and outputs
- 4. Obtaining the program and installation
- 5. Performances, efficiency, parameters
- 5.1. What does impact the memory used by LoRDEC? How much memory do I need to run LoRDEC?
- 5.2. How much time does it take? What does impact the running time?
- 5.3. How is the the de Bruijn Graph built in LoRDEC?
- 5.4. In which language is LoRDEC programmed?
- 5.5. How many cores does LoRDEC use? How can i control the number of cores?
- 5.6. How to choose the value of \(k\)?
- 5.7. What's the meaning of
-aparameter? - 5.8. Can I iterate LoRDEC? Is it worth to iterate LoRDEC?
- 5.9. LoRDEC exhaust my memory, i have too much short read data, how can i do?
- 6. Evaluation of correction
- 7. Miscellaneous
1 Abbreviations
- LR: Long Reads
- SR: Short Reads
- DBG: de Bruijn Graph
- PacBio: Pacific Biosciences
- TP/FP: True Positives / False Positives
- FN : False Negatives
2 Overview and concepts
2.1 Process overview
LoRDEC is an efficient hybrid correction algorithm for long reads.
LoRDEC corrects long reads using a collection of short reads. It works in two steps:
- it reads the short reads and builds a de Bruijn Graph of the short reads.
- Once the graph is built, it reads the long reads one by one, corrects it, and output the corrected long reads.
The de Bruijn Graph depends on a key parameter denoted \(k\).
Remarks:
- The set of long reads can be split into many smaller files and each processed in parallel.
- Once built the graph is saved in a special file for later re-use. This is practical if several long reads files are corrected using the same collection of short reads. See "speeding up" and "parallelisation".
2.2 What is hybrid correction?
Two classes of approaches exist to correct long reads:
- hybrid correction: using short reads to correct long reads
- self correction: using only the information of long reads to correct them. It is an auto-correction in the sense that only the sequence of long reads is used to correct them.
LoRDEC performs hybrid correction.
2.3 Why use hybrid correction?
Hybrid correction takes advantage of the fact that 2nd generation sequencing techniques yields reads that are almost error free compared to long reads. Hence, short read sequences can be trusted. By comparing wrong pieces of long reads with equivalent in short reads one can detects and correct errors in long reads according to short read sequence.
In contrast, self correction requires a large sequencing depth of long read to provide sufficient information for their correction. Getting this sequencing depth remain expensive.
Hybrid correction requires that you have both long reads and a collection of short reads available from the same sample, or the same species. One can often reuse short read data publicly available.
In contrast, self correction requires only long reads.
2.4 Why do we need to correct long reads?
Sequencing technologies of the third generation, like the Single Molecule Real Time (SMRT) sequencing of Pacific Biosciences or MINion from Oxford Nanopore Technologies, generate reads having about 15% to 20% of their positions wrong. Basically, every sixth nucleotide is incorrect. These estimates (provided in 2014 and 2015) depend on the version of the sequencing apparatus and on the chemistry used.
These error rates contrast with those of second generation sequencing, which remain below 1%.
It is difficult to align such raw long reads to a genome for instance, or to compute overlaps between these reads because of the high error rate. Any typical sequence analysis task becomes difficult (i.e. it may fail) and computationally demanding (i.e. it takes long time to run) at that error level. Hence, correction improves the long read sequences and eases their analysis, especially the task of comparison, mapping, and assembly.
2.5 What is a \(k\) -mer?
A \(k\) -mer is a piece of sequence of length \(k\). The value of \(k\) must be an integer.
Consider sequence ATGACTGT which has length 8. If we choose \(k = 3\), then this sequence has \(8-3+1 = 6\) \(k\) -mers: ATG, TGA, GAC, ACT, CTG, TGT.
In a sequence denoted \(S\) (for instance a read), any piece of length \(k\) is a \(k\) -mer. There is one \(k\) -mer starting at almost each position of \(S\) (except those at the very end where there is not enough nucleotides left to make a \(k\) -mer). There are exactly \(\vert S \vert -k +1\) \(k\) -mers. Of course not all \(k\) -mers are necessarily distinct from each other, since any repeat of length larger than \(k\) implies repeated \(k\) -mers.
LoRDEC decomposes the reads into all their \(k\) -mers. This is exactly what the DBG stores in its nodes (see "What is a de Bruijn Graph?").
2.6 What is a de Bruijn Graph?
The de Bruijn Graph of a sequence is a directed graph in the mathematical or computational sense. It is composed of nodes and arcs, which are links between those nodes.
The de Bruijn Graph has a combinatorial origin and definition. But here we mean the assembly de Bruijn Graph, which differs slightly from the graph imagined by the mathematician Nicolaas Govert de Bruijn.
The DBG is the graph underlying most 2nd generation assemblers (e.g., Velvet).
In a DBG, all \(k\) -mers of the sequence (or set of sequences) are represented as nodes, and an arc links a \(k\) -mer to another if the former overlaps the latter by \(k-1\) symbols.
Below, we give an example of DBG for a set of 5 (very) short reads.
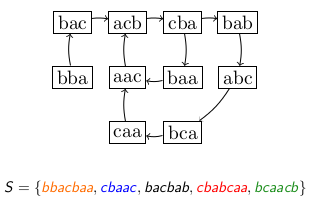
Figure 1: Toy example of a de Bruijn graph for a set of five sequences
2.7 How does LoRDEC use de Bruijn Graph?
In its first step, LoRDEC builds a de Bruijn Graph of the SR. Then, in its second step, it aligns each LR agains similar paths of the DBG.
Note that after correction, LoRDEC saves the DBG into a file in Hierarchical Data Format (HDF) on the disk. By default, the name of this file is the name of the SR data file followed by extension .h5
2.8 Can LoRDEC perform also self-correction?
Not directly. However, LoRDEC has a companion software called LoRMA, that uses LoRDEC and performs self-correction with long reads. LoRMA does not need short reads, but use only long reads.
For the same amount of data, LoRMA requires more computing resources and more time. LoRMA can be downloaded from http://www.cs.helsinki.fi/u/lmsalmel/LoRMA/.
Reference: Accurate selfcorrection of errors in long reads using de Bruijn graphs
L. Salmela, R. Walve, E. Rivals, E. Ukkonen
Bioinformatics, 33 (6): 799-806, 10.1093/bioinformatics/btw321, 2017.
2.9 Can LoRDEC correct transcriptomic long reads?
Yes. The original publication was focused on genomic reads, but LoRDEC can handle both types of assays.
Several large projects have successfully used LoRDEC on whole transcriptome studies. See for instance:
- Global identification of alternative splicing via comparative analysis of SMRT- and Illumina-based RNA-seq in strawberry
Li, Yongping; Dai, Cheng; Hu, Chungen; et al.; Plant Journal 90(1), p. 164-176, DOI: 10.1111/tpj.13462, 2017. - A survey of the sorghum transcriptome using single-molecule long reads
Salah E. Abdel-Ghany, Michael Hamilton, Jennifer L. Jacobi, Peter Ngam, Nicholas Devitt, Faye Schilkey, Asa Ben-Hur & Anireddy S. N. Reddy
Nature Communications 7, Article number: 11706 (2016) 10.1038/ncomms11706 - A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing
Nam V. Hoang, Agnelo Furtado, Patrick J. Mason, Annelie Marquardt, Lakshmi Kasirajan, Prathima P. Thirugnanasambandam, Frederik C. Botha and Robert J. Henry
BMC Genomics 18:395 DOI: 10.1186/s12864-017-3757-8 2017
3 Inputs and outputs
3.1 Input format
LoRDEC accepts multiple formats for the set of short and long reads. The SR can even be "loaded" as a DBG.
3.1.1 Does LoRDEC accepts FastQ or FastA read files?
3.1.2 Does LoRDEC takes compressed read files as input?
Yes. The read files can be compressed with the gzip program. If the original read file is named Illumina.fq then the compressed file usually has the name Illumina.fq.gz
The filename extension .gz usually indicates that the file has been compressed with the gzip compression program.
3.1.3 Input for the de Bruijn Graph.
Instead of the raw SR, you can give as input the de Bruijn graph of these SR that has been created by an earlier execution of LoRDEC on this dataset of SR and for a given k. The datasets and this value of k are encoded in the graph.
This is a file with extension .h5 (unless you have renamed / removed it). See
3.2 Multiple input files. Can i feed LoRDEC with several / multiple input files?
Yes. This makes especially sense for short reads, since they are stored in the graph. For long reads, if you have several files, build the graph once for all and correct each LR file separately. This allows for parallel processing which is faster.
3.3 How do i input several / multiple input files?
The last versions allow multiple Illumina files as input to correct any PacBio library. There are two ways to do that
- a comma separated list of filenames (no blank in between)
- a meta text file containing the list of filenames, one perline.
Each pacbio file can be processed in turn, since each read is processed in turn. So one PacBio file per execution of LoRDEC.
3.3.1 Option 1: Example of correction with multiple short read files
- One PacBio file:
pacbio-mini.fa - Two Illumina files:
ill-test-5K-1.faandill-test-5K-2.fa command for correcting the PacBio file using the two files of Illumina reads:
../lordec-correct -2 ill-test-5K-1.fa,ill-test-5K-2.fa -k 19 -s 3 -i pacbio-mini.fa -o my-corrected-pacbio-reads.fa
Here the two Illumina filenames are given as a single argument after the -2 option: as a single string, where both names are separated by a comma (but no space).
Now if you redirect the standard error to a log file:
../lordec-correct -2 ill-test-5K-1.fa,ill-test-5K-2.fa -k 19 -s 3 -i pacbio-mini.fa -o my-corrected-pacbio-reads.fa &> mylog.log
the message related to the execution of LoRDEC will not appear on your screen.
3.3.2 Option 2: Error correction with multiple short read files
- One PacBio file:
pacbio-mini.fa - Two Illumina files:
ill-test-5K-1.faandill-test-5K-2.fa One file named
meta-file.txtwhich contains two lines like
ill-test-5K-1.fa ill-test-5K-2.fa
command for correcting the PacBio file using the two files of Illumina reads:
lordec-correct -2 meta-file.txt -k 19 -s 3 -i pacbio-mini.fa -o my-corrected-pacbio-reads.fa &> mylog.log
Note that if you used several times the same Illumina sets, you may want to save the graph in a file with a .h5 extension and use it as input in the next correction command.
3.4 Single and paired end short reads
For an explanation about paired end sequencing/reads https://en.wikipedia.org/wiki/Shotgun_sequencing#Paired-end_sequencing.
3.4.1 Does LoRDEC uses paired end reads?
Yes and no. LoRDEC takes advantage of all short reads it has as input. However, it does not use the pairing information.
So in other words, each read is considered as single. LoRDEC does not care whether a read in the first or the second in a pair.
In fact it uses the \(k\) -mer of the short reads, so the pairing information is not exploited in LoRDEC.
3.4.2 How do i give paired end reads to LoRDEC?
The easiest solution is to provide all files as comma separated values. See "Multiple input files".
3.5 What is the file with extenstion .h5?
It contains the graph created by LoRDEC with the SR data. It is a binary file, which is not readable by eye.
This graph is created and stored in a file on the disk either with lordec-correct or with lordec-build-SR-graph commands.
Once it is on the disk, this graph file can be reused for further execution of LoRDEC, provided with you want to use the same set of SR and the same value of k.
4 Obtaining the program and installation
4.1 Getting or using the program
- Official page on the ATGC - NGS web platform: http://www.atgc-montpellier.fr/lordec/
- For a stand alone version on the GATB website: http://gatb.inria.fr/software/lordec/
- For interactive use on a Galaxy platform: http://colibread.genouest.org/
- For a version that you can install in your Galaxy platform: https://github.com/genouest/tools-colibread
4.2 Is LoRDEC working on Galaxy platforms?
Yes. You can download and install it on your Galaxy platform from the Colib'read toolshed: https://github.com/genouest/tools-colibread
4.3 Can use LoRDEC on a Galaxy platform?
Yes. For interactive use on a Galaxy platform see http://colibread.genouest.org/
Thanks to GenOuest by the way.
Reference: Colib'read on Galaxy: A tools suite dedicated to biological information extraction from raw NGS reads
Y. Le Bras, O. Collin, C. Monjeaud, V. Lacroix, E. Rivals, C. Lemaitre, V. Miele, G. Sacomoto, C. Marchet, B. Cazaux, A. Zine El Aabidine, L. Salmela, S. Alves-Carvalho, A. Andrieux, R. Uricaru, P. Peterlongo
GigaScience, Feb 11;5:9. doi: 10.1186/s13742-015-0105-2 eCollection 2016.
4.4 Stand alone version of LoRDEC
The program LoRDEC is written in C++ and uses two external libraries: GATB and Boost.
The version distributed on ATGC - NGS platform needs that these two libraries are installed on your computer. This is quite easy on Linux platform.
However, we also created a stand alone version of LoRDEC, which incorporates GATB library and the parts of Boost library it uses. Hence, the code of the Stand alone version contains all it needs to be compiled and installed. Thus, it is a priori easier to install since you do not need to pre-install neither GATB, nor Boost. The Stand alone version is available as a GATB tools on the GATB core website: http://gatb.inria.fr/software/lordec/.
4.5 Common installation problems
4.5.1 I ran make as instructed in the README but the compiler gives an error message. What should I do?
Check that you are using the correct version of the GATB library. The version number is given in the README file. For the last version of LoRDEC (0.6) you should use GATB Core 1.1.0.
4.5.2 I have the correct version of GATB but LoRDEC still does not compile. What should I do?
LoRDEC uses some newer features in gcc (namely local types as template parameters and lambda expressions). Therefore you need gcc version 4.5 or newer to compile LoRDEC. You can check the version of gcc by running:
g++ --version
If your gcc is older than 4.5, you need to upgrade to a newer version to compile LoRDEC.
5 Performances, efficiency, parameters
5.1 What does impact the memory used by LoRDEC? How much memory do I need to run LoRDEC?
The memory used by LoRDEC depends primarily on the size of the de Bruijn Graph of the short reads.
Fortunately, the graph takes very little space compared to the original size of the short reads data. Actually, the graph is a compressed representation of the original set of short reads.
On maize data comprising 190 millions of short reads, LoRDEC took only 5 gigabases of memory. This correction was done on a laptop.
The long read data has a negligible impact on the memory usage. However it influences the time needed for correction.
5.2 How much time does it take? What does impact the running time?
As each LR is processed one by one, obviously the number of LR, and even more their cumulated lengths, are major determinants of the running time.
Previous to the correction, LoRDEC builds or loads the de Bruijn graph of the short reads. This can also take some time if the SR set is huge.
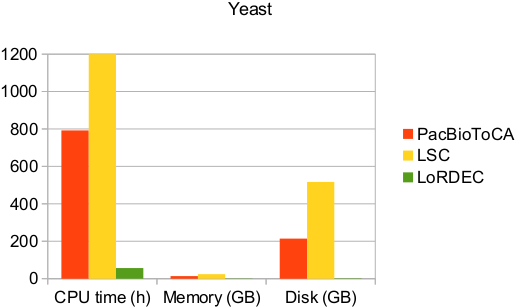
Figure 2: Practical performance on a real yeast data set in terms of running time, of memory and disk usages
5.3 How is the the de Bruijn Graph built in LoRDEC?
5.4 In which language is LoRDEC programmed?
For the sake of efficiency, LoRDEC is programmed in C++, as is the GATB library.
5.5 How many cores does LoRDEC use? How can i control the number of cores?
By default LoRDEC uses all available cores. You can control the number of cores by specifying the parameter -T <number of threads>.
5.6 How to choose the value of \(k\)?
This graph shows acceptable values in quite small range and their effects on the running time (green) and on the quality of correction.

Figure 3: Influence of k-mer length on correction efficiency and running time
Choosing \(k\) is an important issue. Indeed, useful \(k\) -mers for LoRDEC must be shared between the long reads and the short reads. Of course the \(k\) -mer should not be too small: \(k=1\) is obviously inappropriate since sharing a nucleotide is not informative. At the other end, PacBio and Nanopore have many errors that prevent too long \(k\) -mers to be identical between the two read sets. So the size of \(k\) -mer is dictated more by the expected error rate of the long read technology than by the length of reads themselves (short or long), and the smallest \(k\) value allows finding more shared \(k\) -mers. The extreme lower limit is around \(log_4(genome size)\), below which searching for a \(k\) -mer in a random genome of the same size would have almost probability one to be found. So for human like genome, 18 to 20 is reasonable. Below it starts being less informative. We investigated this issue in another context in a paper of NAR (see Fig in there).
Reference:
Using reads to annotate the genome: influence of length, background distribution, and sequence errors on prediction capacity
Philippe, Boureux, Bréhèlin, Tarhio, Commes, Rivals
Nucleic Acids Research (NAR) Vol. 37, No. 15 e104, 10.1093/nar/gkp492; 2009.
5.7 What's the meaning of -a parameter?
It controls the maximum coverage of the k-mers used in the DBG. Indeed, it limits the k-mers to those whose coverage in the short read data is below the maximum value given after -a.
Usage example:
lordec-correct -2 meta-file.txt -k 19 -s 3 -a 10000 -i pacbio-mini.fa -o my-corrected-pacbio-reads.fa &> mylog.log
With command above, the DBG will only comprise the k-mers whose coverage is lower than 10000 (because of -a option) and higher than 3 (because of -s option).
While the option -s controls the minumum abundance for each kmer (for instance -s 3 sets it to 3), the option -a controls the maximum coverage.
Hence you should give a high enough value (say e.g. 100000) following -a, otherwise it may discard many correct kmers.
Optimizing this value depends on your data set. Important questions:
- is it a transcriptomic data set or a genomic one?
- in case of genomic data, what sequencing coverage did you choose in your experimental plan?
5.8 Can I iterate LoRDEC? Is it worth to iterate LoRDEC?
Yes. Inded the amount of correction achieved by LoRDEC depends on the parameter \(k\). So we can cumulate the correction by performing LoRDEC with different \(k\) values.
By iteration, we mean using several times LoRDEC successively, but with increasing values of \(k\). It takes more time, but will improve the correction.
For instance, you may run LoRDEC with \(k=19\), then with \(k=31\), and finally with \(k=41\).
This is how we used it in LoRMA for self correction of long reads. See the paragraph on self-correction.
5.9 LoRDEC exhaust my memory, i have too much short read data, how can i do?
Depending on your hardware, and on the size of your data, LoRDEC may require more memory than available at runtime on your system.
In such case, you may ask LoRDEC to discard more k-mers to only include very sure k-mers in the DBG. This decreases the DBG size and hence the memory needed.
To do so, you can increase slightly the parameter value of parameter -s (using -s 3 instead of -s 2, or using -s 5 instead of -s 3 should already
lordec-correct -2 meta-file.txt -k 19 -s 5 -a 10000 -i pacbio-mini.fa -o my-corrected-pacbio-reads.fa &> mylog.log
Similarly, you may decrease the value following parameter -a.
Alternatively, we have access to computing center on which huge datasets can be processed. Contact us.
6 Evaluation of correction
6.1 Modified Error Correction Toolkit software
An important issue is how results of correction are evualated / assessed. To evaluate LoRDEC, we adapted and used the Error Correction Toolkit (Yang et al., 2013) which was originally designed for comparing error correction results for second-generation sequencing data.
If you need the modified Error Correction Toolkit, for comparing and testing new or existing error correction tools, you can download the archive with the code and scripts either in zip format or in tar.gz format.
These archive files and their content are provided as is, without any garantee. You may use them at your own risks.
Archive content:
- A modified verison of Error Correction Evaluation Toolkit that can handle the local alignments of PacBio reads (it is originally developed for 2nd generation sequencing techs where you get global alignments when mapping).
- ectool-analysis.sh script which you can use to run the evaluation. This assumes that blasr (or bwa if you prefer to use that) is on path and it will also run these. You can take a look at the script to find out the parameters used. Something to note is that if the sam-file for the original reads already exists, the script assumes it is ok to use that (which might not be the case if it was not created with this script but it is handy if you want to evaluate several different correction runs).
- rename-reads.sh script which can be used to rename the reads in the format that Error Correction Evaluation Toolkit assumes. This has to be done before running the error correction tools.
6.2 Information about Error Correction Toolkit
- it requires the mapping of the original reads and of the corrected reads to the genome in SAM format.
- For each pair of original and corrected read, the toolkit computes the set of differences with the reference genome, and it compares these two sets to determine TP, FP and FN positions with regard to correction.
- it computes and reports two statistics:
- Sensitivity = TP/(TP + FN), how well does the tool recognize erroneous positions?
- Gain = (TP − FP)/(TP + FN), how well does the tool remove errors without introducing new ones?
- Sensitivity = TP/(TP + FN), how well does the tool recognize erroneous positions?
- We modified the toolkit so that differences between original and corrected reads are counted only within the genomic region of the local alignment of the original read against the genome.
7 Miscellaneous
7.1 Can i use LoRDEC in a pipeline?
Yes. Several possibilities are offered:
- your own pipeline made using scripts: here LoRDEC is one executables among others. You simply need to install it before.
- you can insert it in a Galaxy pipeline. See the Colib'read toolshed: https://github.com/genouest/tools-colibread
Than LoRDEC can be used before using the long reads data in an assembly pipeline.
7.2 Which publication can I cite?
The algorithm, the software, and its performances are described in
LoRDEC: accurate and efficient long read error correction
L. Salmela and E. Rivals
Bioinformatics 30(24):3506-3514, 2014.
The publication is freely available at 10.1093/bioinformatics/btu538
7.3 Is there a recommended pipeline for correction?
Yes. Here is how we use it. It is important to filter out short reads of lower quality.

Figure 4: Simple pipeline for using LoRDEC
7.4 Limitation on long read length
There is a limit on the size of long read that LoRDEC processes. Currently this limit is set to 500 000 nucleotides.
When it encounters a long read that is longer than this limit, LoRDEC prints an error message ("Too long read") and stops.
#define MAX_READ_LEN 500000
7.4.1 How to change the limit on long read length?
This limits is recorded in the program in the constant called MAX_READ_LEN in file lordec-gen.hpp. One can modify the current value of MAX_READ_LEN and recompile the programs to create new executable programs. This requires that you know how to compile C++ programs. See Installation guide for more details.