LoRMA: a self correction program for long reads (PacBio, Nanopore)
Table of Contents
1 Overview
LoRMA is an error correction program for long reads, which are sequences obtained using the third generation of sequencing technologies (3GS), either with Oxford Nanopore technology or with Pacific Biosciences technology.
LoRMA is a so-called self-correction software, as opposed to e.g. LoRDEC that is a hybrid error correction tool. This means that LoRMA uses only long read sequencing data and thus does not require short read data.
LoRMA proceeds in two phases.
- It iteratively performs local correction of the long reads using LoRDEC (with one special option). The number of LoRDEC iterations is set by the user.
- It then corrects the long read using long-range sequence similarity, which it detects by clustering similar reads using a heuristic multiple alignment procedure.
In the first phase, LoRDEC is run with an increasing parameter \(k\), which defines the \(k\) -mers used in the de Bruijn graph. By default, one performs three runs of LoRDEC correction typically with the k-mer sizes 19, 40 and 61 for a yeast data set.
The more iterations, the better the correction, the longer the execution time.
In the second phase, LoRMA process each read in turn. It searches for other reads that share similar regions with the current read. These similar reads are termed friends. An option controls how many friends are sought. It computes a multiple alignment of this subset of reads and uses the consensus sequence to correct the current read.
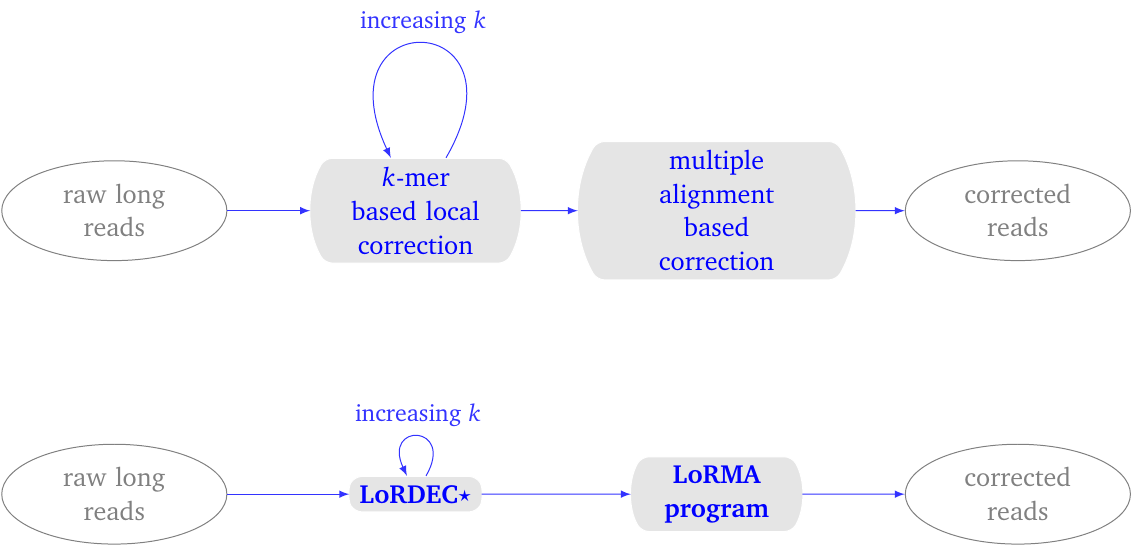
Figure 1: LoRMA process overview. (top) conceptual process. (bottom) pipeline. It uses LoRDEC with a special parameter for self-correction. The entire pipeline can be executed with a single script lorma.sh.
On this site, we provide the program, easy installation procedures (as a linux package or as a conda package), as well as script for parallel execution on large computing servers.
Contact
For comments, support, feedback, requests, etc., please email lorma@lirmm.fr with your name, contact address.
If you wish to be informed about new releases: please send email to sympa@lirmm.fr with a single line:
SUB sub lorma-users your-email@address
remove signature
2 Usage
Calling the program for testing (without doing correction)
If you have installed LoRMA in the directory /home/me/bin/lorma you can simply call the script by typing
/home/me/bin/lorma/lorma.sh
This will print the usage message in your terminal; you should see something alike:
/home/me/bin/lorma/lorma.sh
LoRDEC binaries not found in LoRMA installation directory.
lordec-correct found here: /home/me/bin/miniconda3/bin/lordec-correct
lordec-trim-split found here: /home/me/bin/miniconda3/bin/lordec-trim-split
LoRMA found here: /home/me/bin/lorma/LoRMA
Usage: /home/me/bin/lorma/lorma.sh [-s] [-n] [-start <19> -end <61> -step <21> -threads <6> -friends <7>
-k <19>] [-o OUTPUT_FILE] INPUT_FILE.fasta
-s saves the sequence data of intermediate LoRDEC steps
-n skips LoRDEC steps
Simple example
The long read data file is 10k_lr.fa and it contains 10,000 long reads (of varying length).
lorma.sh -threads 8 10k_lr.fa
performs the whole correction process using 8 cores (if available) on your computer. Finally it yields an output file final.fasta, which contains the corrected and uncorrected reads. You should rename this file before running the process again.
With a modified number of friends
The command
lorma.sh -threads 8 -friends 6 10k_lr.fa
does the same as above but using 6 instead of 7 friends for each read.
3 Frequently Asked Questions (FAQ)
Questions related to error correction in long read, to the underlying concepts (e.g. the de Bruijn graph), are not specific to LoRMA and a lot are answered on lordec FAQ page.
Many options are shared by lordec and LoRMA. Please also take a look at lordec FAQ page.
Abbreviations:
- NGS: Next Generation Sequencing
- 2GS: 2nd Generation Sequencing (i.e., Illumina and other short read technologies)
- 3GS: 3rd Generation Sequencing (i.e., PacBio and Oxford Nanopore long read technologies)
Generalities
What is self correction?
Two kinds of approaches for correcting long reads exist: either hybrid correction or self correction.
- A hybrid method requires also short read data to correct the long reads. Hence, the input of a hybrid method includes short read data files, and long read data files. Such methods (for example lordec) take advantage of the low error rate of short reads to correct long reads.
- A self correction method uses only long reads. It takes advantages of the redundancy in the long read data to find valid sequence information to correct erroneous regions of long reads. This implies that 1/ the input consists of long read data only, and 2/ a higher coverage of long reads is needed (compared to the long read coverage in a hybrid approach).
LoRMA is a self correction method.
- A hybrid method requires also short read data to correct the long reads. Hence, the input of a hybrid method includes short read data files, and long read data files. Such methods (for example lordec) take advantage of the low error rate of short reads to correct long reads.
- How can LoRMA uses lordec for self-correction?
lordec originally is a hybrid error correction tool, and as such, demands as input both a short read datafile and a long read datafile. Here, we twist lordec by giving as input twice the long read datafile. Hence, it will use the \(k\) -mers from the long reads to correct the long reads. However, lordec is used with some special parameters for this sake. - What long read sequencing coverage is needed?
In the experiments on real data performed for the publication, we found that a sequencing coverage 50x is already sufficient. Below that LoRMA can still be used but will tend to correct less regions within the long reads. - What are the uppercase and lowercases in the output file?
Sequence regions in uppercase represent regions with corrected bases, while regions in lowercase are bases that have not been corrected (mostly because, the program could not find enough information, nor consistent information to do so). - Why are my corrected reads shorter than the original ones?
Two reasons. First, during the process, LoRMA trims and splits some of the reads when necessary. Moreover, with Pacific Biosciences reads for instance, errors are predominently insertions, and thus the correction process removes those insertions and shortens the reads. - Can LoRMA correct PacBio (Pacific Biosciences, SMRT) reads?
Yes. We perform extensive tests on real data and some results can be seen in the original publication. - Can LoRMA correct Oxford Nanopore (ONT, MinION, PromethION) reads?
Yes. The way LoRMA processes the long reads is quite general and you can use it to correct ONT reads as well.
We have run extensive tests on Oxford Nanopore data. If you need more information and statistics on ONT data, please contact us. - How can i contact the development team? How can i give feedback?
Please emaillorma@lirmm.frwith your name, contact address and request, comments, support, feedback, etc. - Why are there a program called
LoRMAand a shell script calledlorma.sh?
These are distinct.
- The program
LoRMAis in charge of performing the second phase of the algorithm. - The shell script
lorma.shperforms the whole algorithm by calling programs lordec andLoRMA. This is the script to use. All options can be set using this script.
- The program
Parameters and options
- What is the option friends?
During the second phase, LoRMA searches for other reads that share similar regions with the current read. These similar reads are termed friends. The number of selected friends is a parameter (by default 7). The option-friends xwithxbeing a number allows the user so set the number of friends. The command line
lorma.sh -friends 9 my_lr.fa - What is the option threads?
It allows you to exploit parallelism on your machine. It enables you to use a precise number of cores on your computer or server (which of course depends on your system). So, the command
=lorma.sh -threads 4 my_lr.fa=
uses 4 cores (which shall accelarte the process), while the command without this option
=lorma.sh my_lr.fa=
uses only one (which is slower).
Input format
- Does LoRMA accepts FastQ or FastA read files?
Yes. It accepts both indifferently FastQ and FastA. However, in the current version, LoRMA uses only the ID and sequence information: it does not use the sequence quality information. So giving it FastA or FastQ has the same effect (except that FastQ are of course longer to read).
4 Bibliographic references and links
Original publication in journal Bioinformatics
Accurate self-correction of errors in long reads using de Bruijn graphs
Leena Salmela, Riku Walve Eric Rivals Esko Ukkonen
Bioinformatics, Volume 33, Issue 6, Pages 799–806, 15 March 2017
https://doi.org/10.1093/bioinformatics/btw321
About LoRDEC
5 Acknowledgements and financial support

Figure 2: Current support for maintenance and development of LoRMA

Figure 3: Supports from Finland for the original research and development of LoRMA

Figure 4: Supports from France for the original research and development of LoRMA