
Contact
Please contact me by email for above offers or other enquiries.
( vberry AT lirmm DOT fr )
This is a list of softwares to which I contributed with other authors. Most of them are available on the ATGC bioinformatic platform. Web forms are also available there to run software online on our servers.
Comparing trees
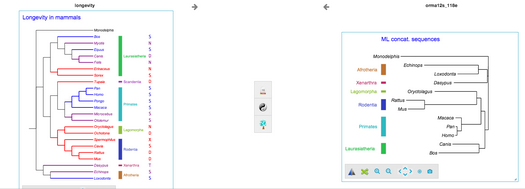
 SylvX is
a reconciliation viewer which implements classical phylogenetic graphic operators (swapping, highlighting, etc.) and new methods to ease interpretation and comparison of reconciliations (multiple maps, moving, shrinking sub-reconciliations).
SylvX is
a reconciliation viewer which implements classical phylogenetic graphic operators (swapping, highlighting, etc.) and new methods to ease interpretation and comparison of reconciliations (multiple maps, moving, shrinking sub-reconciliations).
Reconciliation methods aim at recovering the evolutionary processes that shaped the history of a given gene family including events such as duplications, transfers and losses by comparing the discrepancies between the topologies of the associated gene and species trees. These methods are also used in the framework of host/parasite studies to recover co-diversification scenarios including co-speciation events, host-switches and extinctions. These evolutionary processes can be graphically represented as nested trees. These interconnected graphs can be visually messy and hard to interpret, and despite the fact that reconciliations are increasingly used, there is a shortage of tools dedicated to their graphical management. This is the motivation for proposing SylvX, a tool that allows to play with the graphical representation of a reconciliation. The tool is interfaced with a version of Mowgli, a reconciliator we previously published, but can be used to represent reconciliations proposed from other software (such as Notung and Tera).-
 Mowgli is a command line program that reconciles trees on common taxa sets. It offers numerous options and is specifically time efficient. It provides time consistent scenarii, based on dates proposed for the guide tree (species tree, or host tree). A runtime executable for various operating systems can be found on the ATGC platform.
Mowgli is a command line program that reconciles trees on common taxa sets. It offers numerous options and is specifically time efficient. It provides time consistent scenarii, based on dates proposed for the guide tree (species tree, or host tree). A runtime executable for various operating systems can be found on the ATGC platform.
-
 CompPhy is a web platform to compare gene trees and
comprehensive trees built from supermatrix or supertree inference processes.
It proposes useful functionalities to edit trees (color, root, swap, common signal,...) and
most specifically allows several people to interact live on a common project.
Advanced project management features are also available.
Give it a try at this address.
Personally, I'm extensively using this tool to discuss tree collections with distant collaborators and
it's just fine!
CompPhy is a web platform to compare gene trees and
comprehensive trees built from supermatrix or supertree inference processes.
It proposes useful functionalities to edit trees (color, root, swap, common signal,...) and
most specifically allows several people to interact live on a common project.
Advanced project management features are also available.
Give it a try at this address.
Personally, I'm extensively using this tool to discuss tree collections with distant collaborators and
it's just fine!
(Fiorini et al., to be submitted soon). - ScriptTree a tool to graphically visualize topological differences between trees
and to display annotations wrt parts of phylogenies.
See the Web Site.
(Chevenet et al, Bioinformatics, 2010). - PhyloExplorer a tool to manage and query phylogeny collections and find related published phylogenies. Two versions are available depending on the reference taxonomy:
NCBI or
or ITIS
(Ranwez et al, BMC Evolutionary Biology, 2009). - Program MPC: Multi-Polar Consensus: a program implementing the consensus method described in (Bonnard, Berry & Lartillot, Syst Biol 06) : given a set of phylogenies on the same data set, the idea is to summarize this collection as a set of bipartitions embedded in a small number of trees, rather than in just one tree, as done usually. This allows for the representation of alternative phylogenetic signal otherwise lost due to the compatibility constraint of output bipartitions. Available for download
- Program of the FPT algorithm for computing the Maximum Agreement Subtree (MAST) of rooted trees described in (Berry & Nicolas CPM 04, TCBB 06). Available on request from the first author. See (de Vienne et al 07, von Haeseler et al 08, de Vienne et al 08) for a use of this program in a procedure to obtain a congruency index between two topologies. Associated web site.
- Program of the linear-time 3-approximation algorithm for computing the MAST of rooted trees described in (Berry et al COCOON 05, TALG 09). Available on request from the first author.
Building Supertrees
- SSIMUL a tool to extract signal from multi-gene trees. The idea is to extract speciation signal hidden in those trees among duplication and transfer signal, in order to be able to build the Tree of Life of part of it. See the software page for more details : SSIMUL
- Ancestral Build - v1 and MLS: multi-layer supertree - v0.8 written in Java 1.4. The first is a fast polynomial time algorithm to decide the ancestral compatibility of source trees with internal labels, eg displaying higher-level taxa such as genuses, orders, families. See (Berry & Semple, Syst Biol 06). The program is provided with a graphical interface written with the Jung library that shows the progressive decomposition of the descendancy graph. MLS is a heuristic optimization algorithm to compute a supertree for a set of compatible or incompatible source trees with internal labels (Berry et al, to be submitted). It uses SplitsTree v3 and the MascOpt library. Both Ancestral Build and MLS are available.
- PhySIC: Phylogenetic Signal with Induction and non-Contradiction: a veto supertree method for supertree inference. Online web server. Program also available for download. (Ranwez et al Syst Biol 2007).
- PhySIC_IST: cleaning source trees to infer more informative supertrees: a flexible supertree method that can adopt a broad scale of behaviours from vote to veto when handling topological conflict between input trees. The method also has the advantage of detecting anomalies in each source tree wrt the rest of the collection. This suggests horizontal transfers or reconstruction artefacts such as long branch attraction. Online web server with graphical interface to compare source trees in order to interpret incongruences in the source trees. Program also available for download. (Scornavacca et al BMC Bioinformatics 2008).
Reconstructing phylogenies from quartets
- Several algorithms I contributed with Dave Bryant and Charles Semple are implemented in the current version of SplitsTree: a must-have program in phylogenetics! (Berry & Bryant 99, Bryant & Berry 01, Berry & Semple 06).
- Global Cleaning v1.0 (40k) : correcting wrong quartets. Program of an algorithm in (Berry et al ESA 99) Available for download

Package PHYLOQUART v1.3: exact and heuristic methods (Phylip compatible). Program of the algorithms in (Berry & Gascuel COCOON 96, TCS 00), (Berry & Bryant RECOMB 99, Bryant & Berry Adv Appl Math 01) Available for download