Resources:
A list of different ressources developped during the Phyl-ARIANE project.
Databases
Phyl-ARIANE
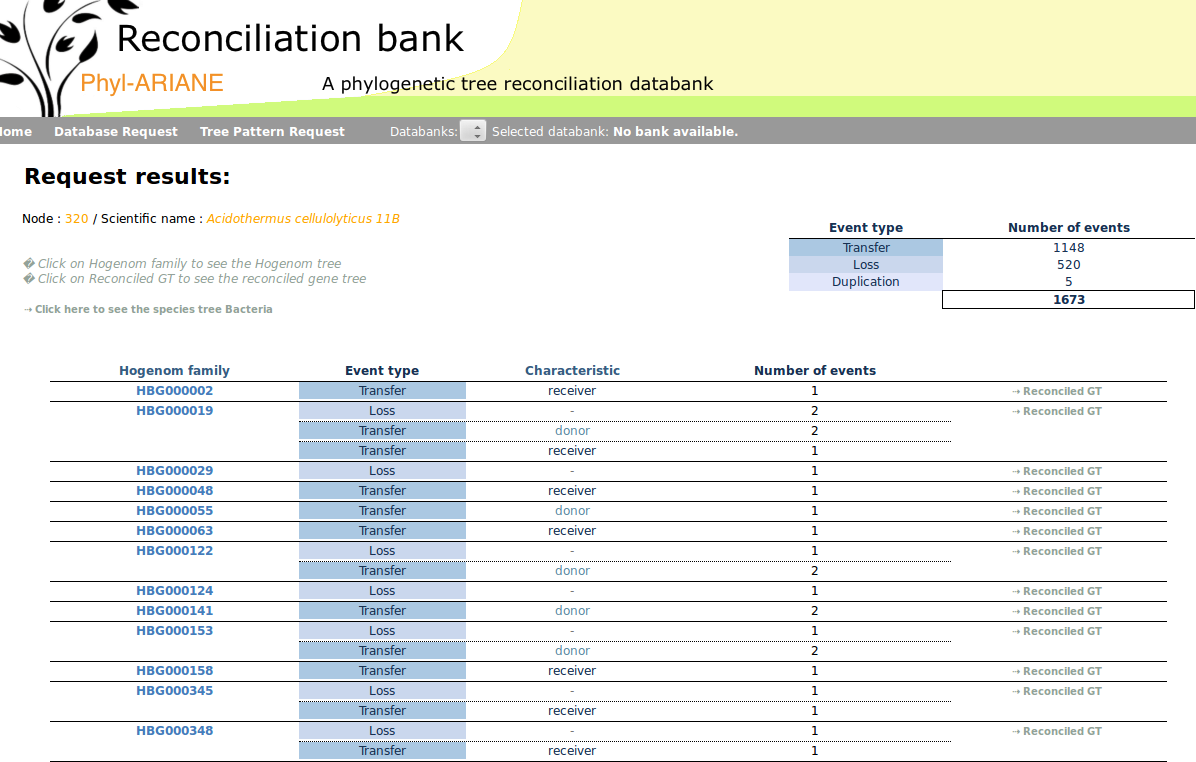
|
Phyl-ARIANE is a phylogenetic tree reconciliation databank. Reconciliations inferred during the project are stored together with a number of important information to be able to trace back the content of ancient genomes, to know the orthology relationships between gene sequences and to detect hot spot of evolution, such as highways of transfers. A first version of the database is publicly available, a v2 is in progress together with new querying and reporting tools (e.g., implementation of the Obirs and Ontofocus tools linked to the Gene Ontology). |
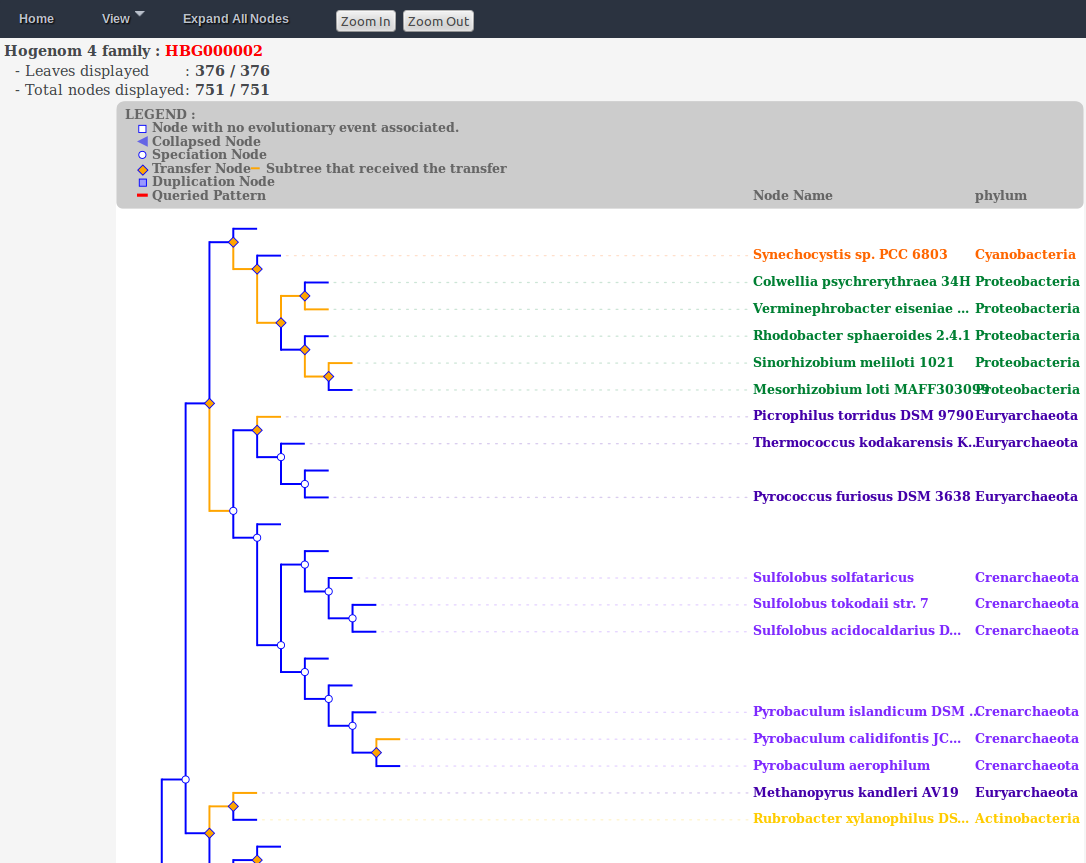
HOGENOM
|
HOGENOM is a database of homologous genes from fully sequenced organisms (bacteria, archeaea and eukarya), structured under ACNUC sequence database management system. It allows to select sets of homologous genes among species, and to visualize multiple alignments and phylogenetic trees. It is as well possible to search for orthologous genes in a wide range of taxons. Thus HOGENOM is particularly useful for comparative sequence analysis, phylogeny and molecular evolution studies. More generaly, HOGENOM gives an overall view of what is known about a peculiar gene family. Note that HOGENOM is splitted into two databases: HOGENOM contains the protein sequences while HOGENOMDNA contains the nucleotide sequences. Protein sequences of HOGENOM have been generated by translating the CDS of HOGENOMDNA and using associated cross-references to generate the annotations. This database pre-existed the Phylariane project but research conducted during the project allowed it to grow in scope and size. The latest release of Hogenom linked to the current project is version 6. |
ORTHOMAM
|
Molecular data play a key role in phylogenetic inference. Mammalian systematics provides us with a clear example, with several previously open evolutionary questions now able to be answered. However, molecular studies have until present used only a handful of classic markers and have not attempted to utilise the information contained within the increasingly large pool of mammalian genome sequences. The identification and utilisation of potentially new informative markers from this pool can help to further resolve the mammalian phylogenetic tree. The EnsEMBL database was used to decide on a set of single-copy orthologous markers from those mammalian genomes available. Exons of reasonable length for further amplification from genomic DNA and sequencing in additional species were then selected. The phylogenetic utility and the evolutionary characteristics of these candidate markers were then evaluated using a homemade bioinformatics pipeline. The resulting OrthoMaM database can be interrogated through this website. The current OrthoMaM release is based on EnsEMBL v54. It now includes 6447 exons and 12958 CDS candidate markers for up to 33 taxa. |
Software
SSIMUL
|
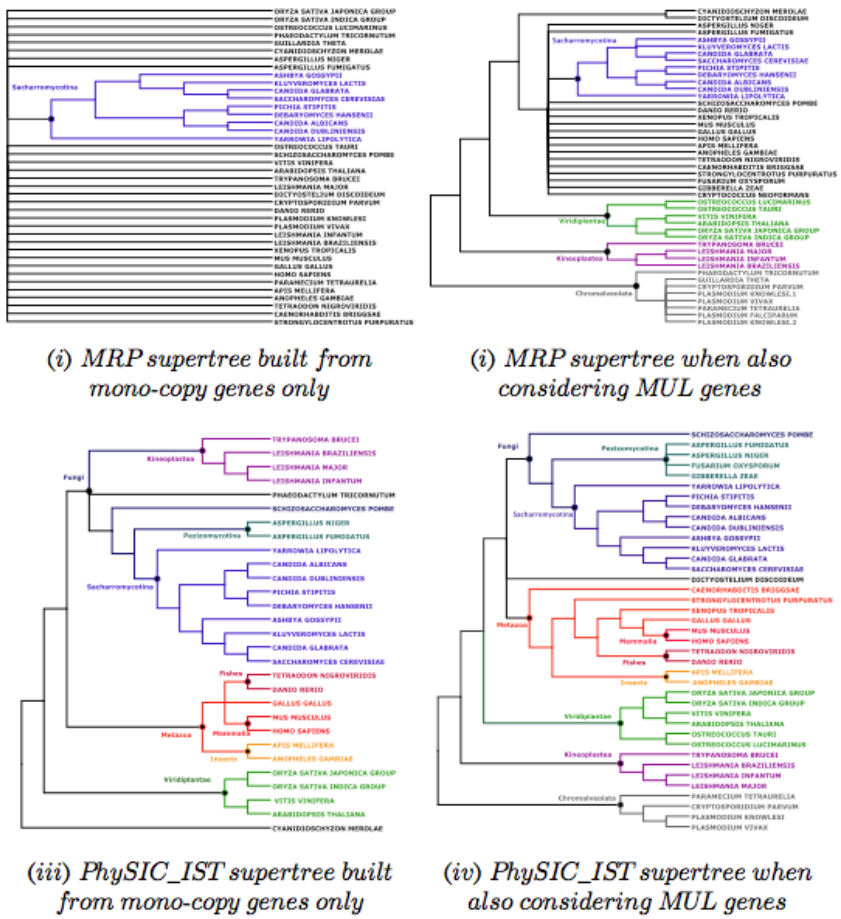
Speciation SIgnal extraction from MULtigene families. Gene trees are leaf-labeled trees inferred from molecular sequences. Because of gene duplication events arising in genomes, some species host several copies of the same gene, hence individual gene trees usually have several leaves labeled with identical species names. Dealing with such multi-labeled gene trees (MUL trees) is a substantial problem in phylogenomics, e.g. current supertree methods do not handle MUL trees, which restricts studies aimed at building the Tree of Life to a very small core of mono-copy genes. We propose to tackle this problem by mainly transforming a collection of MUL trees into a collection of trees, each containing single copies of labels. To achieve that aim, we provide several fast algorithmic building stones. |
Mowgli (MPR) - MowgliNNI
|
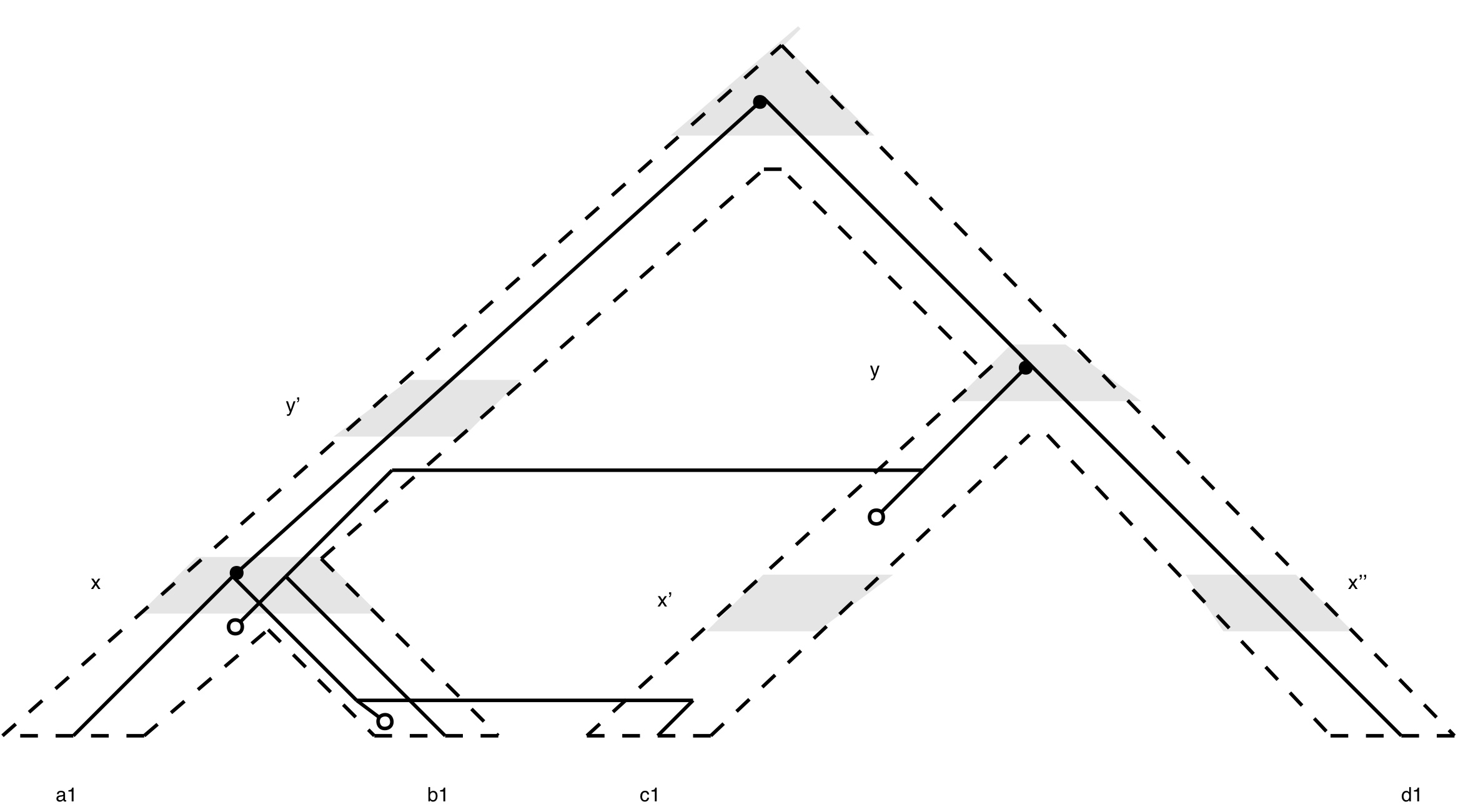
Mowgli (MPR): A fast tree reconciliation program including time consistent transfers, duplications and losses. Tree reconciliation is a computational approach to explain the discrepancy between a gene and a species tree (or between a parasite and a host tree) by postulating a number of evolutionary events such as speciations, duplications, transfers and losses. This approach has important applications in ecology and biogeography but also in genomics to estimate evolutionary scenarios that shaped the content of contemporary genomes and to infer orthology relationships between sequences. MPR is a fast and exact algorithm for reconciling a gene tree and a dated species tree. It optimizes a parsimony criterion according to a model that incorporates all the above-mentioned events (speciation, duplication, loss and transfer). In our experimental study (based on simulated data), parsimony is shown to be an effective criterion to recover the macro-evolutionary events in realistic zones of the parameter space. MowgliNNI: A fast tree reconciliation program considering gene tree uncertainty in duplication-transfer-loss model. MowgliNNI is a variant of Mowgli to reconcile a gene tree with a species tree while accounting for uncertainties in the gene tree. MowgliNNI takes into consideration that gene trees can be partly erroneous due to sequence alignment problems or phylogenetic reconstruction artifacts such as long branch attraction. Thus, it applies Nearest Neighbor Interchange (NNI) edit operations on the edges whose support is lower than a user-chosen threshold to search for a gene tree rearrangement that minimizes the total reconciliation cost according to a given binary species tree. |
RapGreen
|
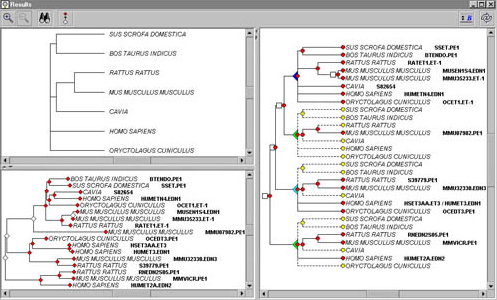
RAP-Green is a new implementation and an improvment of the RAP software, which permits to compare gene and species trees, infers duplication events, and provide confidence score in function conservation between genes. Afterwards, it became a development plateform, to implement several phylogenetic tree analysis softwares. |
Prunier

|
Detecting lateral gene transfers by statistical reconciliation of phylogenetic forests. |
ANGES
|
Reconstructing ANcestral GEnomeS maps. ANGES is a suite of Python programs that allows to reconstruct ancestral genome maps from the comparison of the organization of extant related genomes. ANGES can reconstruct ancestral genome maps for multichromosomal linear genomes and unichromosomal circular genomes. It implements methods inspired from techniques developed to compute physical maps of extant genomes. Examples of cereal, amniote, yeast or bacteria ancestral genomes are provided, computed with ANGES. |
SuperTriplets

|
Phylogenetic tree-building methods use molecular data to describe the evolutionary relationships among genes and taxa, and to infer biodiversity evolutionary patterns. A recurrent problem is to combine the various phylogenies built from different genomic sequences into a single one. This task is generally conducted by a two-step approach whereby a binary matrix representation (MR) of the initial trees is first inferred and then a Maximum Parsimony (MP) analysis is performed on it. This binary representation uses a decomposition of all source trees that is usually based on clades, but that can also be based on triplets or quartets. we focused on the triplet-based representation of source trees and introduce SuperTriplets, a new algorithm that is specially designed to optimize this alternative formulation of the MP criterion. The method avoids several practical limitations of the triplet-based binary matrix representation, making it useful to deal with large datasets. |
PhySIC_IST
|
|
Supertree methods combine phylogenies with overlapping sets of taxa into a larger one. Topological conflicts frequently arise among source trees for methodological or biological reasons, such as long branch attraction, lateral gene transfers, gene duplication/loss or deep gene coalescence. When topological conflicts occur among source trees, liberal methods infer supertrees containing the most frequent alternative, while veto methods infer supertrees not contradicting any source tree, i.e. discard all conflicting resolutions. When the source trees host a significant number of topological conflicts or have a small taxon overlap, supertree methods of both kinds can propose poorly resolved, hence uninformative, supertrees. To overcome this problem, PhySIC_IST propose to infer non-plenary supertrees, i.e. supertrees that do not necessarily contain all the taxa present in the source trees, discarding those whose position greatly differs among source trees or for which insufficient information is provided. |
ScripTree
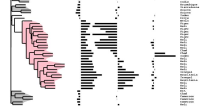
|
There is a large amount of tools for interactive display of phylogenetic trees. However, there is a shortage of tools for the automation of tree rendering. Scripting phylogenetic graphics would enable the saving of graphical analyses involving numerous and complex tree handling operations and would allow the automation of repetitive tasks. ScripTree is a tool intended to fill this gap. It is an interpreter to be used in batch mode. Phylogenetic graphics instructions are stored in a text file and performed in a sequential way. Such instructions are related to tree rendering as well as tree annotation. ScriptTree is written in Tcl/Tk making it a cross-platform application, e.g. suitable for Windows and Unix-like systems, including OS X. It can be used either as a standalone package or included in a bioinformatic pipeline and linked to a HTTP server. |
Generators - Building level-k generators
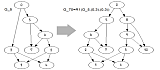
|
Level-k generators were introduced by van Iersel and his coauthors in their RECOMB 2008 paper to describe the structure of simple level-k networks. A case analysis to build all level-2 generators is presented in the detailed version of this article, and later generalized as a brute force algorithm by Steven Kelk followed by a digraph isomorphism test to build all 65 level-3 generators. We generalized these works in showing how general level-k networks can be decomposed into generators of level at most k and provided rules to build level-k+1 generators recursively from level-k generators. A program to automatically build generators is available for download. |
Dendroscope - Galled Networks
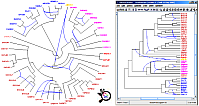
|
In collaboration with Daniel Huson and Regula Rupp, we proposed a new method for building phylogenetic networks from clusters contained in rooted phylogenetic trees. This method is implemented in Dendroscope, a software that is able to visualize rooted phylogenetic trees and networks on thousands of taxa efficiently, and at the same time contains algorithmic machinery to compute consensus trees and rooted phylogenetic networks from a set of trees. |
Treecloud

|
In collaboration with Jean Véronis, we developed a tool to create tree clouds to represent textual data. This new visualization is based on phylogenetic trees and extends tag clouds by representing the most important words of a text with different sizes and colors, around a tree which reflects their proximity in the text. TreeCloud is a free software written in Python. |
Web sites
Querying reconciliations
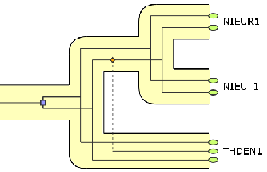
|
This website is an alpha version of the reconciliation bank browser, that will be one of the final production of the Phyl-ARIANE project. The main goal is to provide interactive tools in order to query a databank of evolutionary events: gene duplications, gene losses and horizontal gene transfers. The underlying database of events is currently divided into events concerning two separate groups: gamma and beta proteobacteria. After having chosen a set of target species and having graphically located some macro-evolutionary events (amongst transfer, duplication, losses), the website can be used to query the database thanks to tree pattern matching algorithms. |
PhyloExplorer
| PhyloExplorer is a tool to facilitate assessment and management of phylogenetic tree collections. Given an input collection of rooted trees, PhyloExplorer provides facilities for obtaining statistics describing the collection, correcting invalid taxon names, extracting taxonomically relevant parts of the collection using a dedicated query language, and identifying related trees in the TreeBASE database. |
Phylogenetic Networks

| This website is a database of articles about Phylogenetic Networks. |
MACSE
MACSE (Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons) is the first automatic solution to align protein-coding gene datasets containing non-functional sequences (pseudogenes) without disrupting the underlying codon structure. It has also proved useful in detecting undocumented frameshifts in public database sequences and in aligning next-generation sequencing reads/contigs against a reference coding sequence. |
Ontofocus - OBIRS

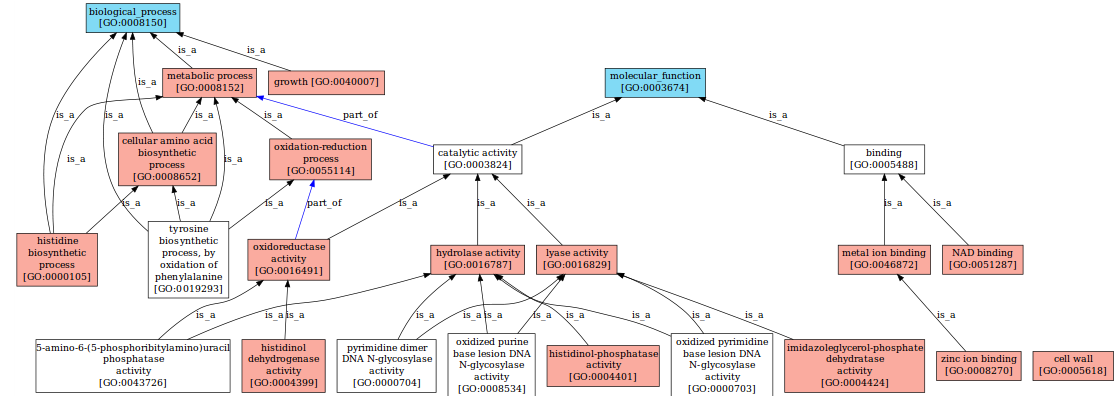
|
Given a reference-ontology, a "good" sub-ontology may be defined as the smallest self-explanatory excerpt containing concepts of interest. Given an initial set of concepts of interest, their relationships can generally not be explicit without adding some hyponyms and hyperonyms. This problem is tackled by using an algorithmic approach based on the is-a relation and two common operators: least common ancestor (lca) and greatest common descendant (gcd). OntoFocus can for instance be used to restrict the Gene Ontology (containing thousands of concepts) to the subset of concepts related to the annotation of the BRCA1 gene (associated with breast cancer susceptibility). OBIRS is an Ontological Based Information Retrieval System, designed to favor user interaction. OBIRS is a request method and an environment based on aggregating models to assess the relevance of documents annotated by concepts of ontology. The selection of documents is displayed in a semantic map to provide graphical indications that make explicit to what extent they match the user’s query; this man/machine interface favors a more interactive and iterative exploration of data corpus, by facilitating the weighting of request concept and visual explanation |
CompPhy
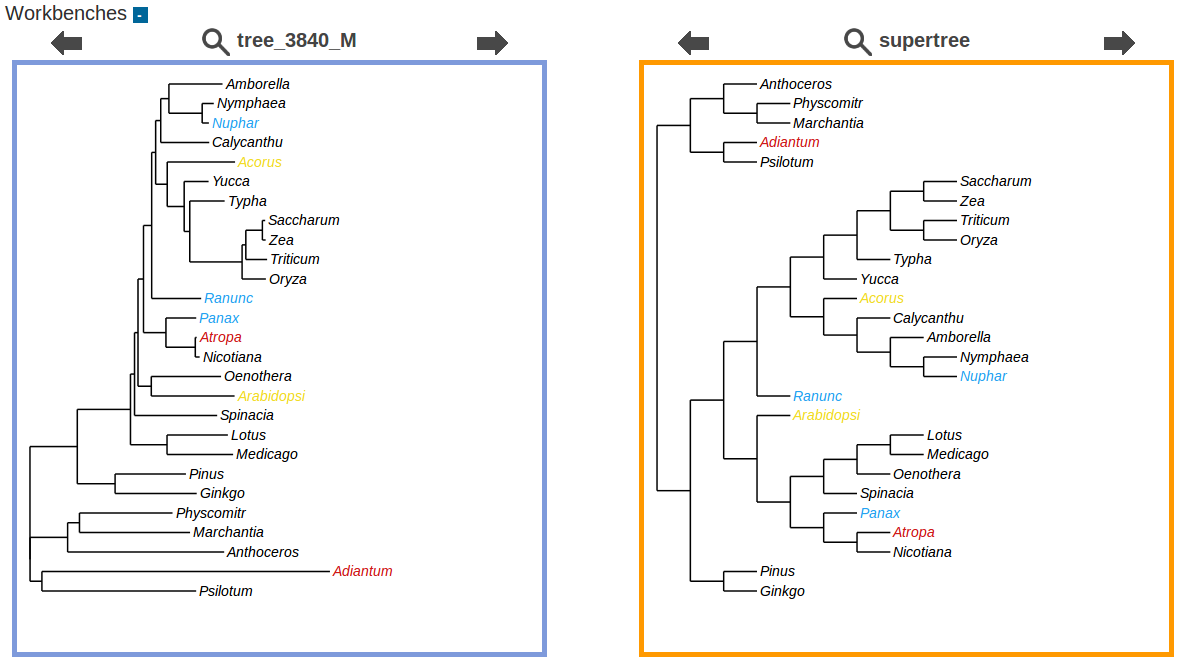
|
CompPhy is a collaborative web-based platform that allows distant people to work together on a phylogeny collection, and perform simple tree comparisons, manipulations and analyses, in a synchronous or asynchronous way. CompPhy is first designed to allow several distant coworkers to jointly manipulate phylogenies, but you can also use it as a personal phylogeny repository, storing trees that awaits for your personal analyses. Last, CompPhy is a way to store phylogenies in the internet "cloud", that is they will be accessible from any computer/internet device you happen to find (after a logging step). In this way, you can carry on the analysis of your trees indifferently from the place you are in. |
