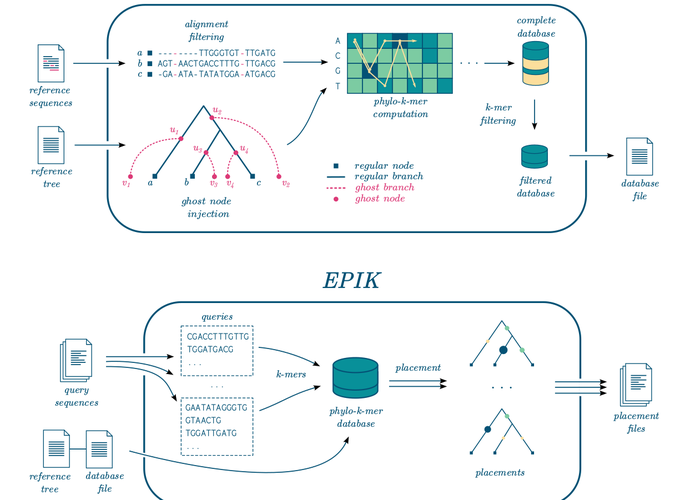
Motivation: Phylogenetic placement enables phylogenetic analysis of massive collections of newly sequenced DNA, when de novo tree inference is too unreliable or inefficient. Assuming that a high-quality reference tree is available, the idea is to seek the correct placement of the new sequences in that tree. Recently, alignment-free approaches to phylogenetic placement have emerged, both to circumvent the need to align the new sequences and to avoid the calculations that typically follow the alignment step. A promising approach is based on the inference of k-mers that can be potentially related to the reference sequences, also called phylo-k-mers. However, its usage is limited by the time and memory-consuming stage of reference data preprocessing and the large numbers of k-mers to consider. Results: We suggest a filtering method for selecting informative phylo-k-mers based on mutual information, which can significantly improve the efficiency of placement, at the cost of a small loss in placement accuracy. This method is implemented in IPK, a new tool for computing phylo-k-mers that significantly outperforms the software previously available. We also present EPIK, a new software for phylogenetic placement, supporting filtered phylo-k-mer databases. Our experiments on real-world data show that EPIK is the fastest phylogenetic placement tool available, when placing hundreds of thousands and millions of queries while still providing accurate placements. Availability and Implementation: IPK and EPIK are freely available at https://github.com/phylo42/IPK and https://github.com/phylo42/EPIK. Both are implemented in C++ and Python and supported on Linux and MacOS. Contact: nromashchenko@lirmm.fr or rivals@lirmm.fr