Counting Overlapping Pairs of Words
A correlation is a binary vector that encodes all possible positions of overlaps of two words, where an overlap for an ordered pair of words (u, v) occurs if a suffix of u matches a prefix of v. As multiple pairs can have the same correlation, it is relevant to count how many pairs of words share the same correlation, depending on the alphabet size and word length n. We exhibit recurrences to compute the number of such pairs – which is termed population size – for any correlation; for this, we exploit a relationship between overlaps of two words and self-overlap of one word. This theorem allows us to compute the number of pairs with the longest overlap of a given length, solving two open questions Gabric raised in 2022. Finally, we also provide bounds for the asymptotic population ratio of any correlation. Given the importance of word overlaps in areas like combinatorics on words, bioinformatics, and digital communication, our results may ease analyses of algorithms for string processing, code design, or genome assembly.

Algorithms to reconstruct past indels: The deletion-only parsimony problem
Ancestral sequence reconstruction is an important task in bioinformatics, with applications ranging from protein engineering to the study of genome evolution. When sequences can only undergo substitutions, optimal reconstructions can be efficiently computed using well-known algorithms. However, accounting for indels in ancestral reconstructions is much harder. First, for biologically-relevant problem formulations, no polynomial-time exact algorithms are available. Second, multiple reconstructions are often equally parsimonious or likely, making it crucial to correctly display uncertainty in the results. Here, we consider a parsimony approach where only deletions are allowed, while addressing the aforementioned limitations. First, we describe an exact algorithm to obtain all the optimal solutions. The algorithm runs in polynomial time if only one solution is sought. Second, we show that all possible optimal reconstructions for a fixed node can be represented using a graph computable in polynomial time. While previous studies have proposed graph-based representations of ancestral reconstructions, this result is the first to offer a solid mathematical justification for this approach. Finally we provide arguments for the relevance of the deletion-only case for the general case.

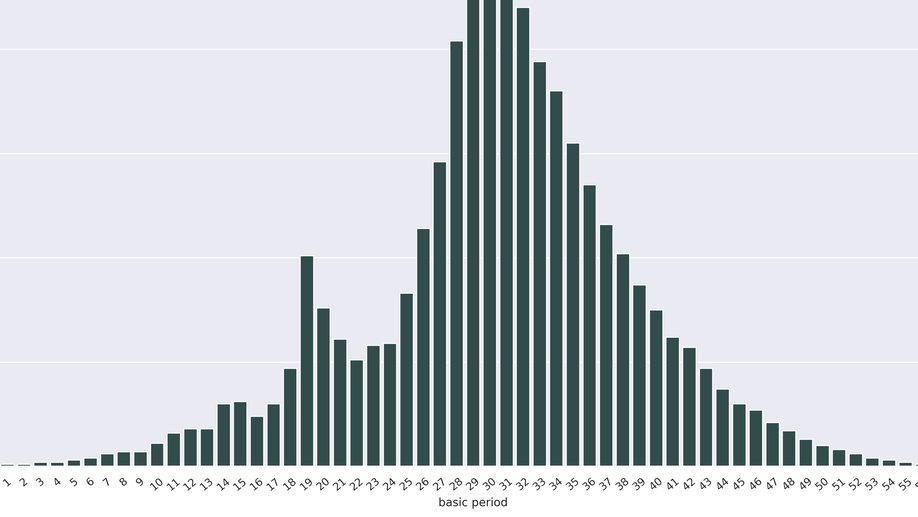
Convergence of the Number of Period sets in Strings
Consider words of length $n$. The set of all periods of a word of length $n$ is a subset of $0,1,2,łdots,n-1$. However, not every subset of $0,1,2,łdots,n-1$ can be a valid set of periods. In a seminal paper in 1981, Guibas and Odlyzko proposed encoding the set of periods of a word into a binary string of length $n$, called an autocorrelation, where a $1$ at position $i$ denotes the period $i$. They considered the question of recognizing a valid period set, and also studied the number $ąppa_n$ of valid period sets for strings of length $n$. They conjectured that $łn p̨pa_n$ asymptotically converges to a constant times $(łn n)^2$. Although improved lower bounds for $łn kp̨a_n/(łn n)^2$ were proved in 2001, the question of a tight upper bound has remained open since Guibas and Odlyzko’s paper. Here, we exhibit an upper bound for this fraction, which implies its convergence and closes this longstanding conjecture. Moreover, we extend our result to find similar bounds for the number of correlations: a generalization of autocorrelations that encodes the overlaps between two strings.
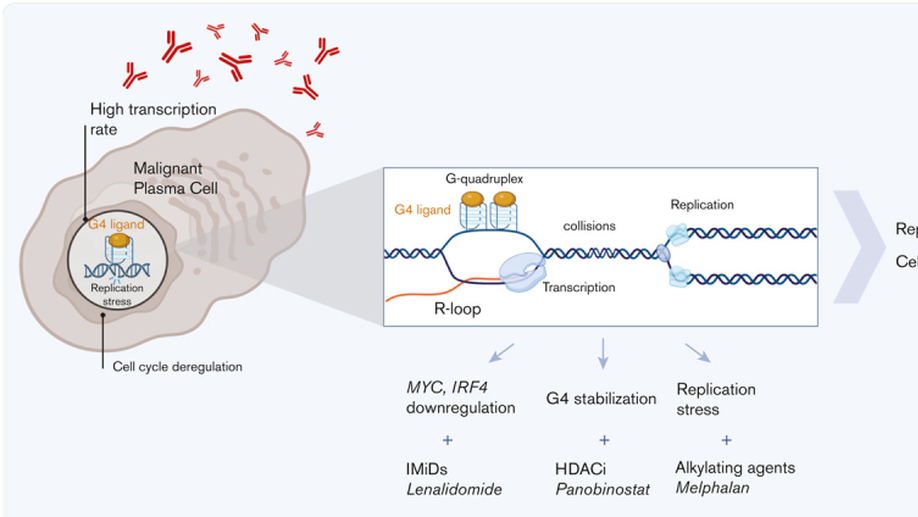
Targeting transcription-replication conflicts using G-quadruplexes stabilizers in multiple myeloma
Replication stress exerts an important role in fueling genomic instability characterizing multiple myeloma (MM) evolution and is a leading cause of drug resistance. Normal and malignant plasma cells (PCs) are associated with a high transcriptional stress due to the huge production of immunoglobulins. Transcription-replication conflicts (TRCs), arising from collisions between replication and transcription machineries, can promote tumor progression and represent an Achilles’ heel to cancer cells. We reported a gene signature related to TRCs management (TRC score), overexpressed in malignant vs normal PCs. High TRC score identified patients with MM with a poor prognosis who could benefit from a TRC-enhancing therapy, in independent cohorts of patients with MM treated with high-dose melphalan chemotherapy or anti-CD38 immunotherapy. Here, we investigated the therapeutic interest of increasing TRCs to target specifically malignant PCs using the G-quadruplex (G4) stabilizer pyridostatin (PDS). PDS exerted significant toxicity in MM cell lines and primary MM cells, inducing DNA damage, cell cycle arrest, and apoptosis. Importantly, primary myeloma cells are significantly more sensitive to PDS treatment than normal bone marrow cells. Moreover, PDS improved the efficacy of MM treatments such as melphalan and histone deacetylase (HDAC) or bromodomain (BRD) inhibitors. Thus, our study shows that G4 stabilizers could be used to specifically target MM cells that exhibit concomitant replication stress and a high level of transcription, through the increase of TRCs. These molecules could be used to increase the efficacy of other treatments including melphalan, HDAC inhibitors, and BRD inhibitors.
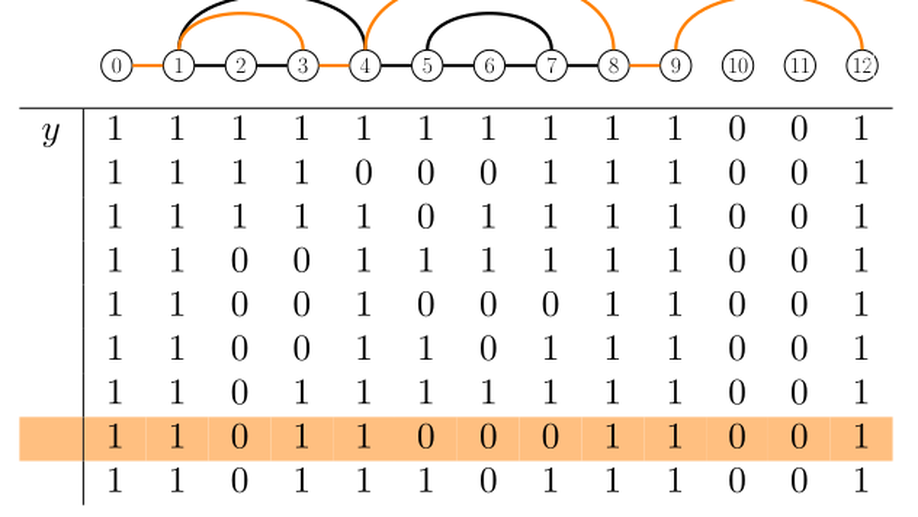
Incremental Computation of the Set of Period Sets
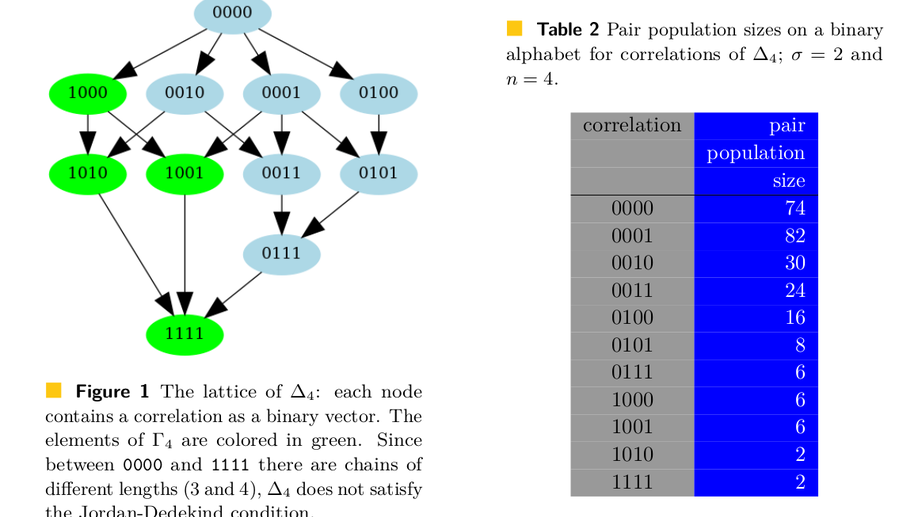
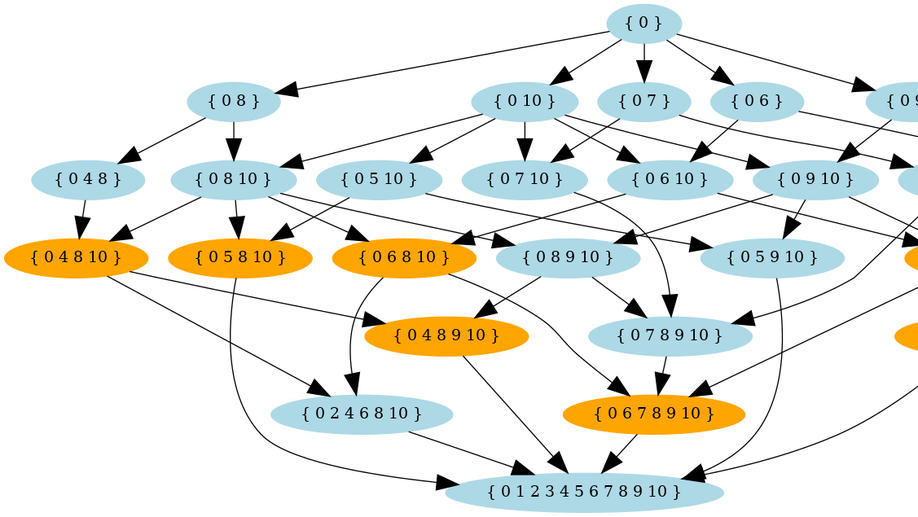
Overlaps between words are crucial in many areas of computer science, such as code design, stringology, and bioinformatics. A self overlapping word is characterized by its periods and borders. A period of a word u is the starting position of a suffix of u that is also a prefix u, and such a suffix is called a border. Each word of length, say n>0, has a set of periods, but not all combinations of integers are sets of periods. Computing the period set of a word u takes linear time in the length of u. We address the question of computing, the set, denoted $Γ_n$, of all period sets of words of length n. Although period sets have been characterized, there is no formula to compute the cardinality of $Γ_n$ (which is exponential in n), and the known dynamic programming algorithm to enumerate $Γ_n$ suffers from its space complexity. We present an incremental approach to compute $Γ_n$ from $Γ_n-1$, which reduces the space complexity, and then a constructive certification algorithm useful for verification purposes. The incremental approach defines a parental relation between sets in $Γ_n-1$ and $Γ_n$, enabling one to investigate the dynamics of period sets, and their intriguing statistical properties. Moreover, the period set of a word u is key for computing the absence probability of u in random texts. Thus, knowing $Γ_n$ is useful to assess the significance of word statistics, such as the number of missing words in a random text.
Algorithms to reconstruct past indels: the deletion-only parsimony problem
Ancestral sequence reconstruction is an important task in bioinformatics, with applications ranging from protein engineering to the study of genome evolution. When sequences can only undergo substitutions, optimal reconstructions can be efficiently computed using well-known algorithms. However, accounting for indels in ancestral reconstructions is much harder. First, for biologically-relevant problem formulations, no polynomial-time exact algorithms are available. Second, multiple reconstructions are often equally parsimonious or likely, making it crucial to correctly display uncertainty in the results. Here, we consider a parsimony approach where any indel event has the same cost, irrespective of its size or the branch where it occurs. We thoroughly examine the case where only deletions are allowed, while addressing the aforementioned limitations. First, we describe an exact algorithm to obtain all the optimal solutions. The algorithm runs in polynomial time if only one solution is sought. Second, we show that all possible optimal reconstructions for a fixed node can be represented using a graph computable in polynomial time. While previous studies have proposed graph-based representations of ancestral reconstructions, this result is the first to offer a solid mathematical justification for this approach. Finally we discuss the relevance of the deletion-only case for the general case.Competing Interest StatementThe authors have declared no competing interest.
Translational control of cell plasticity drives 5-FU tolerance
All routine clinical treatments for colorectal cancer include 5-fluorouracil (5-FU), which cannot counteract recurrence and metastases formation. As the pyrimidine analog 5-FU can impact multiple pathways including both DNA and RNA metabolism, studying its mode of actions could lead to improved therapies. Using a dedicated reporter system for lineage-tracing and deep translatome profiling we demonstrate that 5-FU causes some colorectal cancer cells to tolerate the drug, due to a durable translational reprogramming that sustains cell plasticity. This period of drug tolerance coincides with specific translational activation of genes coding for proteins with major pro-tumoral functions. We unravel a major unexpected translational overexpression of the pro-inflammatory and pro-tumoral IL-8 cytokine, alongside other anti-apoptotic, senescence-associated secretory phenotype and cancer-related senescence phenotype genes. Given the adverse prognostic implications of elevated IL-8 levels across various cancers, our findings suggest IL-8 targeting could counteract 5-FU resistance.
Counting overlapping pairs of strings
A correlation is a binary vector that encodes all possible positions of overlaps of two words, where an overlap for an ordered pair of words (u,v) occurs if a suffix of word u matches a prefix of word v. As multiple pairs can have the same correlation, it is relevant to count how many pairs of words share the same correlation depending on the alphabet size and word length n. We exhibit recurrences to compute the number of such pairs – which is termed population size – for any correlation; for this, we exploit a relationship between overlaps of two words and self-overlap of one word. This theorem allows us to compute the number of pairs with a longest overlap of a given length and to show that the expected length of the longest border of two words asymptotically diverges, which solves two open questions raised by Gabric in 2022. Finally, we also provide bounds for the asymptotic of the population ratio of any correlation. Given the importance of word overlaps in areas like word combinatorics, bioinformatics, and digital communication, our results may ease analyses of algorithms for string processing, code design, or genome assembly.
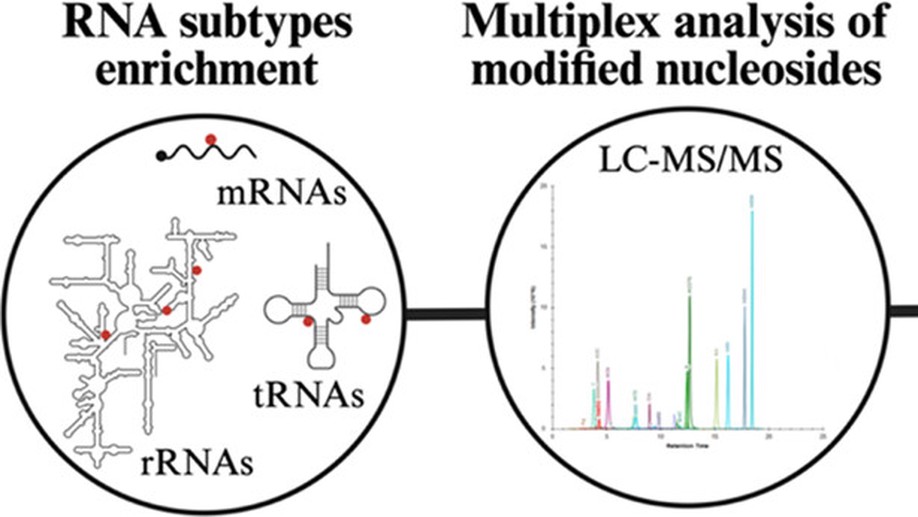
Mass Spectrometry-Based Pipeline for Identifying RNA Modifications Involved in a Functional Process: Application to Cancer Cell Adaptation
Cancer onset and progression are known to be regulated by genetic and epigenetic events, including RNA modifications (a.k.a. epitranscriptomics). So far, more than 150 chemical modifications have been described in all RNA subtypes, including messenger, ribosomal, and transfer RNAs. RNA modifications and their regulators are known to be implicated in all steps of post-transcriptional regulation. The dysregulation of this complex yet delicate balance can contribute to disease evolution, particularly in the context of carcinogenesis, where cells are subjected to various stresses. We sought to discover RNA modifications involved in cancer cell adaptation to inhospitable environments, a peculiar feature of cancer stem cells (CSCs). We were particularly interested in the RNA marks that help the adaptation of cancer cells to suspension culture, which is often used as a surrogate to evaluate the tumorigenic potential. For this purpose, we designed an experimental pipeline consisting of four steps: (1) cell culture in different growth conditions to favor CSC survival; (2) simultaneous RNA subtype (mRNA, rRNA, tRNA) enrichment and RNA hydrolysis; (3) the multiplex analysis of nucleosides by LC-MS/MS followed by statistical/bioinformatic analysis; and (4) the functional validation of identified RNA marks. This study demonstrates that the RNA modification landscape evolves along with the cancer cell phenotype under growth constraints. Remarkably, we discovered a short epitranscriptomic signature, conserved across colorectal cancer cell lines and associated with enrichment in CSCs. Functional tests confirmed the importance of selected marks in the process of adaptation to suspension culture, confirming the validity of our approach and opening up interesting prospects in the field.
Incremental computation of the set of period sets
Overlaps between words are crucial in many areas of computer science, such as code design, stringology, and bioinformatics. A self overlapping word is characterized by its periods and borders. A period of a word $u$ is the starting position of a suffix of $u$ that is also a prefix $u$, and such a suffix is called a border. Each word of length, say $n>0$, has a set of periods, but not all combinations of integers are sets of periods. Computing the period set of a word $u$ takes linear time in the length of $u$. We address the question of computing, the set, denoted $Γ_n$, of all period sets of words of length $n$. Although period sets have been characterized, there is no formula to compute the cardinality of $Γ_n$ (which is exponential in $n$), and the known dynamic programming algorithm to enumerate $Γ_n$ suffers from its space complexity. We present an incremental approach to compute $Γ_n$ from $Γ_n-1$, which reduces the space complexity, and then a constructive certification algorithm useful for verification purposes. The incremental approach defines a parental relation between sets in $Γ_n-1$ and $Γ_n$, enabling one to investigate the dynamics of period sets, and their intriguing statistical properties. Moreover, the period set of a word $u$ is the key for computing the absence probability of $u$ in random texts. Thus, knowing $Γ_n$ is useful to assess the significance of word statistics, such as the number of missing words in a random text.
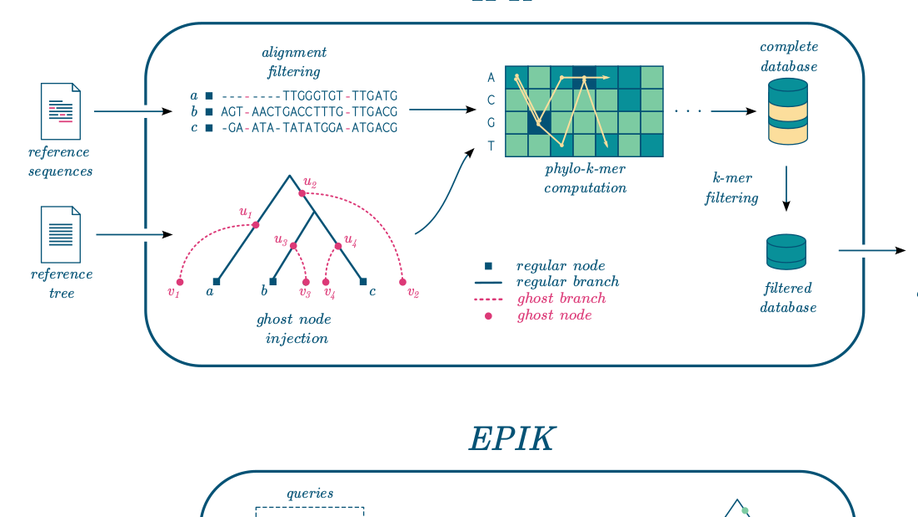
EPIK: Precise and scalable evolutionary placement with informative k-mers
Motivation: Phylogenetic placement enables phylogenetic analysis of massive collections of newly sequenced DNA, when de novo tree inference is too unreliable or inefficient. Assuming that a high-quality reference tree is available, the idea is to seek the correct placement of the new sequences in that tree. Recently, alignment-free approaches to phylogenetic placement have emerged, both to circumvent the need to align the new sequences and to avoid the calculations that typically follow the alignment step. A promising approach is based on the inference of k-mers that can be potentially related to the reference sequences, also called phylo-k-mers. However, its usage is limited by the time and memory-consuming stage of reference data preprocessing and the large numbers of k-mers to consider. Results: We suggest a filtering method for selecting informative phylo-k-mers based on mutual information, which can significantly improve the efficiency of placement, at the cost of a small loss in placement accuracy. This method is implemented in IPK, a new tool for computing phylo-k-mers that significantly outperforms the software previously available. We also present EPIK, a new software for phylogenetic placement, supporting filtered phylo-k-mer databases. Our experiments on real-world data show that EPIK is the fastest phylogenetic placement tool available, when placing hundreds of thousands and millions of queries while still providing accurate placements. Availability and Implementation: IPK and EPIK are freely available at https://github.com/phylo42/IPK and https://github.com/phylo42/EPIK. Both are implemented in C++ and Python and supported on Linux and MacOS. Contact: nromashchenko@lirmm.fr or rivals@lirmm.fr

Physical modeling of ribosomes along messenger RNA: Estimating kinetic parameters from ribosome profiling experiments using a ballistic model
Gene expression is the synthesis of proteins from the information encoded on DNA. One of the two main steps of gene expression is the translation of messenger RNA (mRNA) into polypeptide sequences of amino acids. Here, by taking into account mRNA degradation, we model the motion of ribosomes along mRNA with a ballistic model where particles advance along a filament without excluded volume interactions. Unidirectional models of transport have previously been used to fit the average density of ribosomes obtained by the experimental ribo-sequencing (Ribo-seq) technique in order to obtain the kinetic rates. The degradation rate is not, however, accounted for and experimental data from different experiments are needed to have enough parameters for the fit. Here, we propose an entirely novel experimental setup and theoretical framework consisting in splitting the mRNAs into categories depending on the number of ribosomes from one to four. We solve analytically the ballistic model for a fixed number of ribosomes per mRNA, study the different regimes of degradation, and propose a criterion for the quality of the inverse fit. The proposed method provides a high sensitivity to the mRNA degradation rate. The additional equations coming from using the monosome (single ribosome) and polysome (arbitrary number) ribo-seq profiles enable us to determine all the kinetic rates in terms of the experimentally accessible mRNA degradation rate.
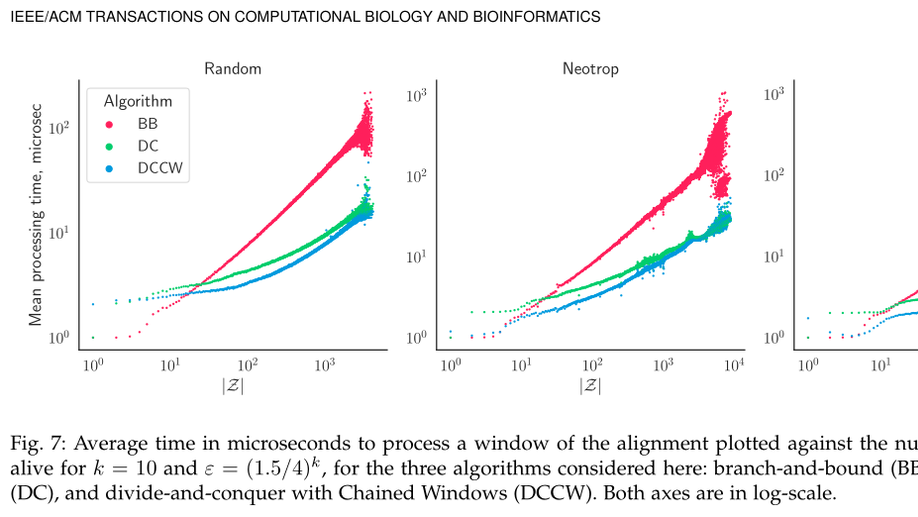
Computing Phylo-$k$-Mers
Finding the correct position of new sequences within an established phylogenetic tree is an increasingly relevant problem in evolutionary bioinformatics and metagenomics. Recently, alignment-free approaches for this task have been proposed. One such approach is based on the concept of phylogenetically-informative k-mers or phylo- k-mers for short. In practice, phylo- k-mers are inferred from a set of related reference sequences and are equipped with scores expressing the probability of their appearance in different locations within the input reference phylogeny. Computing phylo- k-mers, however, represents a computational bottleneck to their applicability in real-world problems such as the phylogenetic analysis of metabarcoding reads and the detection of novel recombinant viruses. Here we consider the problem of phylo- k-mer computation: how can we efficiently find all k-mers whose probability lies above a given threshold for a given tree node? We describe and analyze algorithms for this problem, relying on branch-and-bound and divide-and-conquer techniques. We exploit the redundancy of adjacent windows of the alignment to save on computation. Besides computational complexity analyses, we provide an empirical evaluation of the relative performance of their implementations on simulated and real-world data. The divide-and-conquer algorithms are found to surpass the branch-and-bound approach, especially when many phylo- k-mers are found.

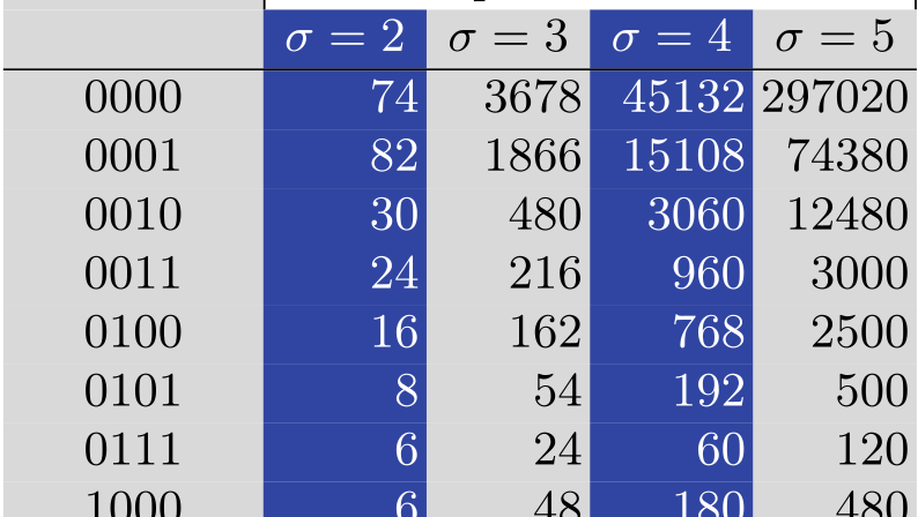
Periodicity of Degenerate Strings
The notion of periods is key in stringology, word combinatorics, and pattern matching algorithms. A string has period p if every two letters at distance p from each other are equal. There has been a growing interest in more general models of sequences which can describe uncertainty. An important model of sequences with uncertainty are degenerate strings. A degenerate string is a string with “undetermined” symbols, which can denote arbitrary subsets of the alphabet $Σ$. Degenerate strings have been extensively used to describe uncertainty in DNA, RNA, and protein sequences using the IUPAC code (Biochemistry, 1970). In this work, we extend the work of Blanchet-Sadri et al. (2010) to obtain the following results about the combinatorial aspects of periodicity for degenerate strings: - We compare three natural generalizations of periodicity for degenerate strings, which we refer to as weak, medium and strong periodicity. We define the concept of total autocorrelations, which are quaternary vectors indicating these three notions of periodicity. - We characterize the three families of period sets, as well as the family of total autocorrelations, for each alphabet size. In particular, we prove necessary conditions period sets should satisfy and, to prove sufficiency, we show how to construct a degenerate string which gives rise to particular period sets. - For each notion of periodicity, we (asymptotically) count the number of period sets, by combining known techniques from partial words with recent results from number theory. - Moreover, we show that all families of period sets, as well as the family of total autocorrelations, form lattices under a suitably defined partial ordering. - We compute the population of weak, medium and strong period sets (i.e., the number of strings with that period set). We also compute the population of total autocorrelations.
dipwmsearch: a Python package for searching di-PWM motifs
Seeking probabilistic motifs in a sequence is a common task to annotate putative transcription factor binding sites or other RNA/DNA binding sites. Useful motif representations include position weight matrices (PWMs), dinucleotide PWMs (di-PWMs), and hidden Markov models (HMMs). Dinucleotide PWMs not only combine the simplicity of PWMs—a matrix form and a cumulative scoring function—but also incorporate dependency between adjacent positions in the motif (unlike PWMs which disregard any dependency). For instance to represent binding sites, the HOCOMOCO database provides di-PWM motifs derived from experimental data. Currently, two programs, SPRy-SARUS and MOODS, can search for occurrences of di-PWMs in sequences.We propose a Python package called dipwmsearch, which provides an original and efficient algorithm for this task (it first enumerates matching words for the di-PWM, and then searches these all at once in the sequence, even if the latter contains IUPAC codes). The user benefits from an easy installation via Pypi or conda, a comprehensive documentation, and executable scripts that facilitate the use of di-PWMs.dipwmsearch is available at https://pypi.org/project/dipwmsearch/ and https://gite.lirmm.fr/rivals/dipwmsearch/ under Cecill license.
FT-GPI, a highly sensitive and accurate predictor of GPI-anchored proteins, reveals the composition and evolution of the GPI proteome in Plasmodium species
Background: Protozoan parasites are known to attach specific and diverse group of proteins to their plasma membrane via a GPI anchor. In malaria parasites, GPI-anchored proteins (GPI-APs) have been shown to play an important role in host-pathogen interactions and a key function in host cell invasion and immune evasion. Because of their immunogenic properties, some of these proteins have been considered as malaria vaccine candidates. However, identification of all possible GPI-APs encoded by these parasites remains challenging due to their sequence diversity and limitations of the tools used for their characterization. Methods: The FT-GPI software was developed to detect GPI-APs based on the presence of a hydrophobic helix at both ends of the premature peptide. FT-GPI was implemented in C ++and applied to study the GPI-proteome of 46 isolates of the order Haemosporida. Using the GPI proteome of Plasmodium falciparum strain 3D7 and Plasmodium vivax strain Sal-1, a heuristic method was defined to select the most sensitive and specific FT-GPI software parameters. Results: FT-GPI enabled revision of the GPI-proteome of P. falciparum and P. vivax, including the identification of novel GPI-APs. Orthology- and synteny-based analyses showed that 19 of the 37 GPI-APs found in the order Haemosporida are conserved among Plasmodium species. Our analyses suggest that gene duplication and deletion events may have contributed significantly to the evolution of the GPI proteome, and its composition correlates with speciation. Conclusion: FT-GPI-based prediction is a useful tool for mining GPI-APs and gaining further insights into their evolution and sequence diversity. This resource may also help identify new protein candidates for the development of vaccines for malaria and other parasitic diseases. Keywords: FT-GPI; GPI-anchored protein; GPI-proteome; P. vivax; Plasmodium falciparum.
Convergence of the Number of Period Sets in Strings
Consider words of length n. The set of all periods of a word of length n is a subset of 0,1,2,…,n-1. However, any subset of 0,1,2,…,n-1 is not necessarily a valid set of periods. In a seminal paper in 1981, Guibas and Odlyzko proposed to encode the set of periods of a word into an n long binary string, called an autocorrelation, where a one at position i denotes the period i. They considered the question of recognizing a valid period set, and also studied the number of valid period sets for strings of length n, denoted κ_n. They conjectured that ln(κ_n) asymptotically converges to a constant times ln²(n). Although improved lower bounds for ln(κ_n)/ln²(n) were proposed in 2001, the question of a tight upper bound has remained open since Guibas and Odlyzko’s paper. Here, we exhibit an upper bound for this fraction, which implies its convergence and closes this longstanding conjecture. Moreover, we extend our result to find similar bounds for the number of correlations: a generalization of autocorrelations which encodes the overlaps between two strings.
Multivariate Analysis of RNA Chemistry Marks Uncovers Epitranscriptomics-Based Biomarker Signature for Adult Diffuse Glioma Diagnostics
One of the main challenges in cancer management relates to the discovery of reliable biomarkers, which could guide decision-making and predict treatment outcome. In particular, the rise and democratization of high-throughput molecular profiling technologies bolstered the discovery of “biomarker signatures” that could maximize the prediction performance. Such an approach was largely employed from diverse OMICs data (i.e., genomics, transcriptomics, proteomics, metabolomics) but not from epitranscriptomics, which encompasses more than 100 biochemical modifications driving the post-transcriptional fate of RNA: stability, splicing, storage, and translation. We and others have studied chemical marks in isolation and associated them with cancer evolution, adaptation, as well as the response to conventional therapy. In this study, we have designed a unique pipeline combining multiplex analysis of the epitranscriptomic landscape by high-performance liquid chromatography coupled to tandem mass spectrometry with statistical multivariate analysis and machine learning approaches in order to identify biomarker signatures that could guide precision medicine and improve disease diagnosis. We applied this approach to analyze a cohort of adult diffuse glioma patients and demonstrate the existence of an “epitranscriptomics-based signature” that permits glioma grades to be discriminated and predicted with unmet accuracy. This study demonstrates that epitranscriptomics (co)evolves along cancer progression and opens new prospects in the field of omics molecular profiling and personalized medicine.
The multifaceted functions of the Fat mass and Obesity-associated protein (FTO) in normal and cancer cells
The last decade has seen mRNA modification emerge as a new layer of gene expression regulation. The Fat mass and obesity-associated protein (FTO) was the first identified eraser of N6-methyladenosine (m6A) adducts, the most widespread modification in eukaryotic messenger RNA. This discovery, of a reversible and dynamic RNA modification, aided by recent technological advances in RNA mass spectrometry and sequencing has led to the birth of the field of epitranscriptomics. FTO crystallized much of the attention of epitranscriptomics researchers and resulted in the publication of numerous, yet contradictory, studies describing the regulatory role of FTO in gene expression and central biological processes. These incongruities may be explained by a wide spectrum of FTO substrates and RNA sequence preferences: FTO binds multiple RNA species (mRNA, snRNA and tRNA) and can demethylate internal m6A in mRNA and snRNA, N6,2′-O-dimethyladenosine (m6Am) adjacent to the mRNA cap, and N1-methyladenosine (m1A) in tRNA. Here, we review current knowledge related to FTO function in healthy and cancer cells. In particular, we emphasize the divergent role(s) attributed to FTO in different tissues and subcellular and molecular contexts.
Physical modeling of ribosomes along messenger RNA: estimating kinetic parameters from ribosome profiling experiments using a ballistic model
Gene expression consists in the synthesis of proteins from the information encoded on DNA. One of the two main steps of gene expression is the translation of messenger RNA (mRNA) into polypeptide sequences of amino acids. Here, by taking into account mRNA degradation, we model the motion of ribosomes along mRNA with a ballistic model where particles advance along a filament without excluded volume interactions. Unidirectional models of transport have previously been used to fit the average density of ribosomes obtained by the experimental ribo-sequencing (Ribo-seq) technique. In this case an inverse fit gives access to the kinetic rates: the position-dependent speeds and the entry rate of ribosomes onto mRNA. The degradation rate is not, however, accounted for and experimental data from different experiments are needed to have enough parameters for the fit. Here, we propose an entirely novel experimental setup and theoretical framework consisting in splitting the mRNAs into categories depending on the number of ribosomes from one to four. We solve analytically the ballistic model for a fixed number of ribosomes per mRNA, study the different regimes of degradation, and propose a criteria for the quality of the inverse fit. The proposed method provides a high sensitivity to the mRNA degradation rate. The additional equations coming from using the monosome (single ribosome) and polysome (arbitrary number) ribo-seq profiles enable us to determine all the kinetic rates in terms of the experimentally accessible mRNA degradation rate.
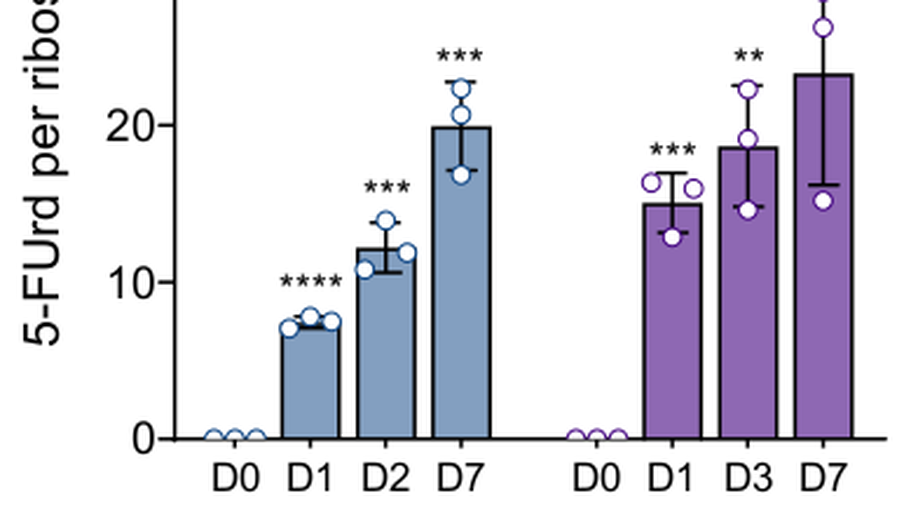
Alteration of ribosome function upon 5-fluorouracil treatment favors cancer cell drug-tolerance
Mechanisms of drug-tolerance remain poorly understood and have been linked to genomic but also to non-genomic processes. 5-fluorouracil (5-FU), the most widely used chemotherapy in oncology is associated with resistance. While prescribed as an inhibitor of DNA replication, 5-FU alters all RNA pathways. Here, we show that 5-FU treatment leads to the production of fluorinated ribosomes exhibiting altered translational activities. 5-FU is incorporated into ribosomal RNAs of mature ribosomes in cancer cell lines, colorectal xenografts, and human tumors. Fluorinated ribosomes appear to be functional, yet, they display a selective translational activity towards mRNAs depending on the nature of their 5′-untranslated region. As a result, we find that sustained translation of IGF-1R mRNA, which encodes one of the most potent cell survival effectors, promotes the survival of 5-FU-treated colorectal cancer cells. Altogether, our results demonstrate that “man-made” fluorinated ribosomes favor the drug-tolerant cellular phenotype by promoting translation of survival genes.
FTO-mediated cytoplasmic m6Am demethylation adjusts stem-like properties in colorectal cancer cell
Cancer stem cells (CSCs) are a small but critical cell population for cancer biology since they display inherent resistance to standard therapies and give rise to metastases. Despite accruing evidence establishing a link between deregulation of epitranscriptome-related players and tumorigenic process, the role of messenger RNA (mRNA) modifications in the regulation of CSC properties remains poorly understood. Here, we show that the cytoplasmic pool of fat mass and obesity-associated protein (FTO) impedes CSC abilities in colorectal cancer through its N6,2’-O-dimethyladenosine (m6Am) demethylase activity. While m6Am is strategically located next to the m7G-mRNA cap, its biological function is not well understood and has not been addressed in cancer. Low FTO expression in patient-derived cell lines elevates m6Am level in mRNA which results in enhanced in vivo tumorigenicity and chemoresistance. Inhibition of the nuclear m6Am methyltransferase, PCIF1/CAPAM, fully reverses this phenotype, stressing the role of m6Am modification in stem-like properties acquisition. FTO-mediated regulation of m6Am marking constitutes a reversible pathway controlling CSC abilities. Altogether, our findings bring to light the first biological function of the m6Am modification and its potential adverse consequences for colorectal cancer management.
A Linear Time Algorithm for Constructing Hierarchical Overlap Graphs
The hierarchical overlap graph (HOG) is a graph that encodes overlaps from a given set P of n strings, as the overlap graph does. A best known algorithm constructs HOG in O(||P|| log n) time and O(||P||) space, where ||P|| is the sum of lengths of the strings in P. In this paper we present a new algorithm to construct HOG in O(||P||) time and space. Hence, the construction time and space of HOG are better than those of the overlap graph, which are O(||P|| + n²).
Rapid screening and detection of inter-type viral recombinants using phylo-k-mers
Novel recombinant viruses may have important medical and evolutionary significance, as they sometimes display new traits not present in the parental strains. This is particularly concerning when the new viruses combine fragments coming from phylogenetically distinct viral types. Here, we consider the task of screening large collections of sequences for such novel recombinants. A number of methods already exist for this task. However, these methods rely on complex models and heavy computations that are not always practical for a quick scan of a large number of sequences.We have developed SHERPAS, a new program to detect novel recombinants and provide a first estimate of their parental composition. Our approach is based on the precomputation of a large database of ‘phylogenetically-informed k-mers’, an idea recently introduced in the context of phylogenetic placement in metagenomics. Our experiments show that SHERPAS is hundreds to thousands of times faster than existing software, and enables the analysis of thousands of whole genomes, or long-sequencing reads, within minutes or seconds, and with limited loss of accuracy.The source code is freely available for download at https://github.com/phylo42/sherpas.Supplementary data are available at Bioinformatics online.
Efficient Construction of Hierarchical Overlap Graphs
The hierarchical overlap graph (HOG for short) is an overlap encoding graph that efficiently represents overlaps from a given set $P$ of $n$ strings. A previously known algorithm constructs the HOG in $O(|| P || + n^2)$ time and $O(|| P || +n times min (n,max |s|:s ın P))$ space, where $|| P ||$ is the sum of lengths of the $n$ strings in $P$. We present a new algorithm of $O(|| P || łog n)$ time and $O(|| P || )$ space to compute the HOG, which exploits the segment tree data structure. We also propose an alternative algorithm using $O(|| P || fracłog nłog łog n)$ time and $O(|| P ||)$ space in the word RAM model of computation.
PEWO: a collection of workflows to benchmark phylogenetic placement
Abstract Motivation Phylogenetic placement (PP) is a process of taxonomic identification for which several tools are now available. However, it remains difficult to assess which tool is more adapted to particular genomic data or a particular reference taxonomy. We developed PEWO, the first benchmarking tool dedicated to PP assessment. Its automated workflows can evaluate PP at many levels, from parameter optimisation for a particular tool, to the selection of the most appropriate genetic marker when PP-based species identifications are targeted. Our goal is that PEWO will become a community effort and a standard support for future developments and applications of phylogenetic placement. Availability https://github.com/phylo42/PEWO Supplementary information Supplementary data are available at Bioinformatics online.
Hierarchical Overlap Graph
Given a set of finite words, the Overlap Graph (OG) is a complete weighted digraph where each word is a node and where the weight of an arc equals the length of the longest overlap of one word onto the other (Overlap is an asymmetric notion). The OG serves to assemble DNA fragments or to compute shortest superstrings, which are a compressed representation of the input. The OG requires space that is quadratic in the number of words, which limits its scalability. The Hierarchical Overlap Graph (HOG) is an alternative graph that also encodes all maximal overlaps, but uses space that is linear in the sum of the lengths of the input words. We propose the first algorithm to build the HOG in linear space for words of equal length.
Greedy-reduction from Shortest Linear Superstring to Shortest Circular Superstring
A superstring of a set of strings correspond to a string which contains all the other strings as substrings. The problem of finding the Shortest Linear Superstring is a well-know and well-studied problem in stringology. We present here a variant of this problem, the Shortest Circular Superstring problem where the sought superstring is a circular string. We show a strong link between these two problems and prove that the Shortest Circular Superstring problem is NP-complete. Moreover, we propose a new conjecture on the approximation ratio of the Shortest Circular Superstring problem.
Linking indexing data structures to de Bruijn graphs: Construction and update
DNA sequencing technologies have tremendously increased their throughput, and hence complicated DNA assembly. Numerous assembly programs use de Bruijn graphs (dBG) built from short reads to merge these into contigs, which represent putative DNA segments. In a dBG of order k, nodes are substrings of length k of reads (or k-mers), while arcs are their k+1-mers. As analysing reads often require to index all their substrings, it is interesting to exhibit algorithms that directly build a dBG from a pre-existing index, and especially a contracted dBG, where non-branching paths are condensed into single nodes. Here, we exhibit linear time algorithms for constructing the full or contracted dBGs from suffix trees, suffix arrays, and truncated suffix trees. With the latter the construction uses a space that is linear in the size of the dBG. Finally, we also provide algorithms to dynamically update the order of the graph without reconstructing it.
AQUAPONY: visualization and interpretation of phylogeographic information on phylogenetic trees
Motivation The visualization and interpretation of evolutionary spatiotemporal scenarios is broadly and increasingly used in infectious disease research, ecology or agronomy. Using probabilistic frameworks, well-known tools can infer from molecular data ancestral traits for internal nodes in a phylogeny, and numerous phylogenetic rendering tools can display such evolutionary trees. However, visualizing such ancestral information and its uncertainty on the tree remains tedious. For instance, ancestral nodes can be associated to several geographical annotations with close probabilities and thus, several migration or transmission scenarios exist. Results We expose a web-based tool, named AQUAPONY, that facilitates such operations. Given an evolutionary tree with ancestral (e.g. geographical) annotations, the user can easily control the display of ancestral information on the entire tree or a subtree, and can view alternative phylogeographic scenarios along a branch according to a chosen uncertainty threshold. AQUAPONY interactively visualizes the tree and eases the objective interpretation of evolutionary scenarios. AQUAPONY’s implementation makes it highly responsive to user interaction, and instantaneously updates the tree visualizations even for large trees (which can be exported as image files). Availability and implementation AQUAPONY is coded in JavaScript/HTML, available under Cecill license, and can be freely used at http://www.atgc-montpellier.fr/aquapony/.
NF-Y controls fidelity of transcription initiation at gene promoters through maintenance of the nucleosome-depleted region
Faithful transcription initiation is critical for accurate gene expression, yet the mechanisms underlying specific transcription start site (TSS) selection in mammals remain unclear. Here, we show that the histone-fold domain protein NF-Y, a ubiquitously expressed transcription factor, controls the fidelity of transcription initiation at gene promoters in mouse embryonic stem cells. We report that NF-Y maintains the region upstream of TSSs in a nucleosome-depleted state while simultaneously protecting this accessible region against aberrant and/or ectopic transcription initiation. We find that loss of NF-Y binding in mammalian cells disrupts the promoter chromatin landscape, leading to nucleosomal encroachment over the canonical TSS. Importantly, this chromatin rearrangement is accompanied by upstream relocation of the transcription pre-initiation complex and ectopic transcription initiation. Further, this phenomenon generates aberrant extended transcripts that undergo translation, disrupting gene expression profiles. These results suggest NF-Y is a central player in TSS selection in metazoans and highlight the deleterious consequences of inaccurate transcription initiation.
Linking BWT and XBW via Aho-Corasick Automaton: Applications to Run-Length Encoding
The boom of genomic sequencing makes compression of sets of sequences inescapable. This underlies the need for multi-string indexing data structures that helps compressing the data. The most prominent example of such data structures is the Burrows-Wheeler Transform (BWT), a reversible permutation of a text that improves its compressibility. A similar data structure, the eXtended Burrows-Wheeler Transform (XBW), is able to index a tree labelled with alphabet symbols. A link between a multi-string BWT and the Aho-Corasick automaton has already been found and led to a way to build a XBW from a multi-string BWT. We exhibit a stronger link between a multi-string BWT and a XBW by using the order of the concatenation in the multi-string. This bijective link has several applications: first, it allows one to build one data structure from the other; second, it enables one to compute an ordering of the input strings that optimises a Run-Length measure (i.e., the compressibility) of the BWT or of the XBW.
Fast and Accurate Genome-Scale Identification of DNA-Binding Sites
Discovering DNA binding sites in genome sequences is crucial for understanding genomic regulation. Currently available computational tools for finding binding sites with Position Weight Matrices of known motifs are often used in restricted genomic regions because of their long run times. The ever-increasing number of complete genome sequences points to the need for new generations of algorithms capable of processing large amounts of data. Here we present MOTIF, a new algorithm for seeking transcription factor binding sites in whole genome sequences in a few seconds. We propose a web service that enables the users to search for their own matrix or for multiple JASPAR matrices. Beyond its efficacy, the service properly handles undetermined positions within the genome sequence and provides an adequate output listing for each position the matching word and its score. MOTIF is available through a web interface at http://www.atgc-montpellier.fr/motif.
Relationship between superstring and compression measures: New insights on the greedy conjecture
A superstring of a set of words is a string that contains each input word as a substring. Given such a set, the Shortest Superstring Problem (SSP) asks for a superstring of minimum length. SSP is an important theoretical problem related to the Asymmetric Travelling Salesman Problem, and also has practical applications in data compression and in bioinformatics. Indeed, it models the question of assembling a genome from a set of sequencing reads. Unfortunately, SSP is known to be NP-hard even on a binary alphabet and also hard to approximate with respect to the superstring length or to the compression achieved by the superstring. Even the variant in which all words share the same length r, called r-SSP, is NP-hard whenever r>2. Numerous involved approximation algorithms achieve approximation ratio above 2 for the superstring, but remain difficult to implement in practice. In contrast the greedy conjecture asked in 1988 whether a simple greedy algorithm achieves ratio of 2 for SSP. Here, we present a novel approach to bound the superstring approximation ratio with the compression ratio, which, when applied to the greedy algorithm, shows a 2 approximation ratio for 3-SSP, and also that greedy achieves ratios smaller than 2. This leads to a new version of the greedy conjecture.
Superstrings with multiplicities
A superstring of a set of words $P = s_1, …, s_p $ is a string that contains each word of P as substring. Given P, the well known Shortest Linear Superstring problem (SLS), asks for a shortest superstring of P. In a variant of SLS, called Multi-SLS, each word s_i comes with an integer m(i), its multiplicity, that sets a constraint on its number of occurrences, and the goal is to find a shortest superstring that contains at least m(i) occurrences of s_i. Multi-SLS generalizes SLS and is obviously as hard to solve, but it has been studied only in special cases (with words of length 2 or with a fixed number of words). The approximability of Multi-SLS in the general case remains open. Here, we study the approximability of Multi-SLS and that of the companion problem Multi-SCCS, which asks for a shortest cyclic cover instead of shortest superstring. First, we investigate the approximation of a greedy algorithm for maximizing the compression offered by a superstring or by a cyclic cover: the approximation ratio is 1/2 for Multi-SLS and 1 for Multi-SCCS. Then, we exhibit a linear time approximation algorithm, Concat-Greedy, and show it achieves a ratio of 4 regarding the superstring length. This demonstrates that for both measures Multi-SLS belongs to the class of APX problems.
Practical lower and upper bounds for the Shortest Linear Superstring
Given a set P of words, the Shortest Linear Superstring (SLS) problem is an optimisation problem that asks for a superstring of P of minimal length. SLS has applications in data compression, where a superstring is a compact representation of P, and in bioinformatics where it models the first step of genome assembly. Unfortunately SLS is hard to solve (NP-hard) and to closely approximate (MAX-SNP-hard). If numerous polynomial time approximation algorithms have been devised, few articles report on their practical performance. We lack knowledge about how closely an approximate superstring can be from an optimal one in practice. Here, we exhibit a linear time algorithm that reports an upper and a lower bound on the length of an optimal superstring. The upper bound is the length of an approximate superstring. This algorithm can be used to evaluate beforehand whether one can get an approximate superstring whose length is close to the optimum for a given instance. Experimental results suggest that its approximation performance is orders of magnitude better than previously reported practical values. Moreover, the proposed algorithm remainso efficient even on large instances and can serve to explore in practice the approximability of SLS.
Computational pan-genomics: status, promises and challenges
Many disciplines, from human genetics and oncology to plant breeding, microbiology and virology, commonly face the challenge of analyzing rapidly increasing numbers of genomes. In case of Homo sapiens, the number of sequenced genomes will approach hundreds of thousands in the next few years. Simply scaling up established bioinformatics pipelines will not be sufficient for leveraging the full potential of such rich genomic data sets. Instead, novel, qualitatively different computational methods and paradigms are needed. We will witness the rapid extension of computational pan-genomics, a new sub-area of research in computational biology. In this article, we generalize existing definitions and understand a pan-genome as any collection of genomic sequences to be analyzed jointly or to be used as a reference. We examine already available approaches to construct and use pan-genomes, discuss the potential benefits of future technologies and methodologies and review open challenges from the vantage point of the above-mentioned biological disciplines. As a prominent example for a computational paradigm shift, we particularly highlight the transition from the representation of reference genomes as strings to representations as graphs. We outline how this and other challenges from different application domains translate into common computational problems, point out relevant bioinformatics techniques and identify open problems in computer science. With this review, we aim to increase awareness that a joint approach to computational pan-genomics can help address many of the problems currently faced in various domains.
Functional analysis of Plasmodium falciparum subpopulations associated with artemisinin resistance in Cambodia
Plasmodium falciparum malaria is one of the most widespread parasitic infections in humans and remains a leading global health concern. Malaria elimination efforts are threatened by the emergence and spread of resistance to artemisinin-based combination therapy, the first-line treatment of malaria. Promising molecular markers and pathways associated with artemisinin drug resistance have been identified, but the underlying molecular mechanisms of resistance remains unknown. The genomic data from early period of emergence of artemisinin resistance (2008–2011) was evaluated, with aim to define k13 associated genetic background in Cambodia, the country identified as epicentre of anti-malarial drug resistance, through characterization of 167 parasite isolates using a panel of 21,257 SNPs.
The cacao Criollo genome v2.0: an improved version of the genome for genetic and functional genomic studies
Theobroma cacao L., native to the Amazonian basin of South America, is an economically important fruit tree crop for tropical countries as a source of chocolate. The first draft genome of the species, from a Criollo cultivar, was published in 2011. Although a useful resource, some improvements are possible, including identifying misassemblies, reducing the number of scaffolds and gaps, and anchoring un-anchored sequences to the 10 chromosomes.
Ribo-seq enlightens codon usage bias
Codon usage is biased between lowly and highly expressed genes in a genome-specific manner. This universal bias has been well assessed in some unicellular species, but remains problematic to assess in more complex species. We propose a new method to compute codon usage bias based on genome wide translational data. A new technique based on sequencing of ribosome protected mRNA fragments (Ribo-seq) allowed us to rank genes and compute codon usage bias with high precision for a great variety of species, including mammals. Genes ranking using Ribo-Seq data confirms the influence of the tRNA pool on codon usage bias and shows a decreasing bias in multicellular species. Ribo-Seq analysis also makes possible to detect preferred codons without information on genes function.
Rearrangement Scenarios Guided by Chromatin Structure
Genome architecture can be drastically modified through a succession of large-scale rearrangements. In the quest to infer these rearrangement scenarios, it is often the case that the parsimony principal alone does not impose enough constraints. In this paper we make an initial effort towards computing scenarios that respect chromosome conformation, by using Hi-C data to guide our computations. We confirm the validity of a model – along with optimization problems Minimum Local Scenario and Minimum Local Parsimonious Scenario – developed in previous work that is based on a partition into equivalence classes of the adjacencies between syntenic blocks. To accomplish this we show that the quality of a clustering of the adjacencies based on Hi-C data is directly correlated to the quality of a rearrangement scenario that we compute between Drosophila melanogaster and Drosophila yakuba. We evaluate a simple greedy strategy to choose the next rearrangement based on Hi-C, and motivate the study of the solution space of Minimum Local Parsimonious Scenario.
Population genomics of picophytoplankton unveils novel chromosome hypervariability
Tiny photosynthetic microorganisms that form the picoplankton (between 0.3 and 3 μm in diameter) are at the base of the food web in many marine ecosystems, and their adaptability to environmental change hinges on standing genetic variation. Although the genomic and phenotypic diversity of the bacterial component of the oceans has been intensively studied, little is known about the genomic and phenotypic diversity within each of the diverse eukaryotic species present. We report the level of genomic diversity in a natural population of Ostreococcus tauri (Chlorophyta, Mamiellophyceae), the smallest photosynthetic eukaryote. Contrary to the expectations of clonal evolution or cryptic species, the spectrum of genomic polymorphism observed suggests a large panmictic population (an effective population size of 1.2 × 107) with pervasive evidence of sexual reproduction. De novo assemblies of low-coverage chromosomes reveal two large candidate mating-type loci with suppressed recombination, whose origin may pre-date the speciation events in the class Mamiellophyceae. This high genetic diversity is associated with large phenotypic differences between strains. Strikingly, resistance of isolates to large double-stranded DNA viruses, which abound in their natural environment, is positively correlated with the size of a single hypervariable chromosome, which contains 44 to 156 kb of strain-specific sequences. Our findings highlight the role of viruses in shaping genome diversity in marine picoeukaryotes.
Full Compressed Affix Tree Representations
The Suffix Tree, a crucial and versatile data structure for string analysis of large texts, is often used in pattern matching and in bioinformatics applications. The Affix Tree generalizes the Suffix Tree in that it supports full tree functionalities in both search directions. The bottleneck of Affix Trees is their space requirement for storing the data structure. Here, we discuss existing representations and classify them into two categories: Synchronous and Asynchronous. We design Compressed Affix Tree indexes in both categories and explored how to support all tree operations bidirectionally. This work compares alternative approaches for compressing the Affix Tree, measuring their space and time trade-offs for different operations. Moreover, to our knowledge, this is the first work that compares all Compressed Affix Tree implementations offering a practical benchmark for this structure.
De novo assembly of viral quasispecies using overlap graphs
A viral quasispecies, the ensemble of viral strains populating an infected person, can be highly diverse. For optimal assessment of virulence, pathogenesis, and therapy selection, determining the haplotypes of the individual strains can play a key role. As many viruses are subject to high mutation and recombination rates, high-quality reference genomes are often not available at the time of a new disease outbreak. We present SAVAGE, a computational tool for reconstructing individual haplotypes of intra-host virus strains without the need for a high-quality reference genome. SAVAGE makes use of either FM-index–based data structures or ad hoc consensus reference sequence for constructing overlap graphs from patient sample data. In this overlap graph, nodes represent reads and/or contigs, while edges reflect that two reads/contigs, based on sound statistical considerations, represent identical haplotypic sequence. Following an iterative scheme, a new overlap assembly algorithm that is based on the enumeration of statistically well-calibrated groups of reads/contigs then efficiently reconstructs the individual haplotypes from this overlap graph. In benchmark experiments on simulated and on real deep-coverage data, SAVAGE drastically outperforms generic de novo assemblers as well as the only specialized de novo viral quasispecies assembler available so far. When run on ad hoc consensus reference sequence, SAVAGE performs very favorably in comparison with state-of-the-art reference genome-guided tools. We also apply SAVAGE on two deep-coverage samples of patients infected by the Zika and the hepatitis C virus, respectively, which sheds light on the genetic structures of the respective viral quasispecies.
The power of greedy algorithms for approximating Max-ATSP, Cyclic Cover, and superstrings
The Maximum Asymmetric Travelling Salesman Problem (Max-ATSP), which asks for a Hamiltonian path of maximum weight covering a digraph, and the Maximum Compression(Max-Comp), which, for a finite language $P ≔s1,…,sp$, asks for w, a superstring of P minimising ∑si∈S|si|−|w|, are both difficult to approximate (Max-SNP hard). Solving Max-ATSP on the overlap graph of the words of P solves Max-Comp. Many approximate algorithms have been designed to improve their approximation ratios, but these are increasingly complex. Often, these rely on solving the pendant problems where the cover is made of cycles instead of single path (Max-CC and SCCS). Thus, the greedy algorithm remains an attractive solution for its simplicity and ease of implementation. Here, using the full power of subset systems, we provide a unified approach for proving simply the approximation ratios of a greedy algorithm for these four problems. In addition, we introduce two new problems dealing with the case of cyclic input words, and exhibit a greedy approximation ratio for them. The Maximum Partitioned Hamiltonian Path generalises the Maximum Asymmetric Travelling Salesman Problem when the nodes are partitioned into classes and the path must contain one element of each class. The Maximum Cyclic Compression is the natural counterpart of Maximum Compression for cyclic strings.
Read mapping on de Bruijn graphs
Background. Next Generation Sequencing (NGS) has dramatically enhanced our ability to sequence genomes, but not to assemble them. In practice, many published genome sequences remain in the state of a large set of contigs. Each contig describes the sequence found along some path of the assembly graph, however, the set of contigs does not record all the sequence information contained in that graph. Although many subsequent analyses can be performed with the set of contigs, one may ask whether mapping reads on the contigs is as informative as mapping them on the paths of the assembly graph. Currently, one lacks practical tools to perform mapping on such graphs. Results. Here, we propose a formal definition of mapping on a de Bruijn graph, analyse the problem complexity which turns out to be NP-complete, and provide a practical solution. We propose a pipeline called GGMAP (Greedy Graph MAPping). Its novelty is a procedure to map reads on branching paths of the graph, for which we designed a heuristic algorithm called BGREAT (de Bruijn Graph REAd mapping Tool). For the sake of efficiency, BGREAT rewrites a read sequence as a succession of unitigs sequences. GGMAP can map millions of reads per CPU hour on a de Bruijn graph built from a large set of human genomic reads. Surprisingly, results show that up to 22% more reads can be mapped on the graph but not on the contig set. Conclusions. Although mapping reads on a de Bruijn graph is complex task, our proposal offers a practical solution combining efficiency with an improved mapping capacity compared to assembly-based mapping even for complex eukaryotic data.
Accurate self-correction of errors in long reads using de Bruijn graphs
New long read sequencing technologies, like PacBio SMRT and Oxford NanoPore, can produce sequencing reads up to 50 000 bp long but with an error rate of at least 15%. Reducing the error rate is necessary for subsequent utilization of the reads in, e.g. de novo genome assembly. The error correction problem has been tackled either by aligning the long reads against each other or by a hybrid approach that uses the more accurate short reads produced by second generation sequencing technologies to correct the long reads.We present an error correction method that uses long reads only. The method consists of two phases: first, we use an iterative alignment-free correction method based on de Bruijn graphs with increasing length of k-mers, and second, the corrected reads are further polished using long-distance dependencies that are found using multiple alignments. According to our experiments, the proposed method is the most accurate one relying on long reads only for read sets with high coverage. Furthermore, when the coverage of the read set is at least 75x, the throughput of the new method is at least 20% higher.LoRMA is freely available at http://www.cs.helsinki.fi/u/lmsalmel/LoRMA/.
Shortest DNA Cyclic Cover in Compressed Space
For a set of input words, finding a superstring (a string containing each word of the set as a substring) of minimal length is hard. Most approximation algorithms solve the Shortest Cyclic Cover problem before merging the cyclic strings into a linear superstring. A cyclic cover is a set of cyclic strings in which the input words occur as a substring. We investigate a variant of the Shortest Cyclic Cover problem for the case of DNA. Because the two strands that compose DNA have a reverse complementary sequence, and because the sequencing process often overlooks the strand of a read, each read or its reverse complement must occur as a substring in a cyclic cover. We exhibit a linear time algorithm based on graphs for solving the Shortest DNA Cyclic Cover problem and propose compressed data structures for storing the underlying graphs. All results and algorithms can be adapted to the case where strings are simply reversed but not complemented (e.g. in pattern recognition).
A linear time algorithm for Shortest Cyclic Cover of Strings
Merging words according to their overlap yields a superstring. This basic operation allows to infer long strings from a collection of short pieces, as in genome assembly. To capture a maximum of overlaps, the goal is to infer the shortest superstring of a set of input words. The Shortest Cyclic Cover of Strings (SCCS) problem asks, instead of a single linear superstring, for a set of cyclic strings that contain the words as substrings and whose sum of lengths is minimal. SCCS is used as a crucial step in polynomial time approximation algorithms for the notably hard Shortest Superstring problem, but it is solved in cubic time. The cyclic strings are then cut and merged to build a linear superstring. SCCS can also be solved by a greedy algorithm. Here, we propose a linear time algorithm for solving SCCS based on a Eulerian graph that captures all greedy solutions in linear space. Because the graph is Eulerian, this algorithm can also find a greedy solution of SCCS with the least number of cyclic strings. This has implications for solving certain instances of the Shortest linear or cyclic Superstring problems.
Colib’read on galaxy: a tools suite dedicated to biological information extraction from raw NGS reads
With next-generation sequencing (NGS) technologies, the life sciences face a deluge of raw data. Classical analysis processes for such data often begin with an assembly step, needing large amounts of computing resources, and potentially removing or modifying parts of the biological information contained in the data. Our approach proposes to focus directly on biological questions, by considering raw unassembled NGS data, through a suite of six command-line tools.Dedicated to ‘whole-genome assembly-free’ treatments, the Colib’read tools suite uses optimized algorithms for various analyses of NGS datasets, such as variant calling or read set comparisons. Based on the use of a de Bruijn graph and bloom filter, such analyses can be performed in a few hours, using small amounts of memory. Applications using real data demonstrate the good accuracy of these tools compared to classical approaches. To facilitate data analysis and tools dissemination, we developed Galaxy tools and tool shed repositories.With the Colib’read Galaxy tools suite, we enable a broad range of life scientists to analyze raw NGS data. More importantly, our approach allows the maximum biological information to be retained in the data, and uses a very low memory footprint.
Superstring Graph: A New Approach for Genome Assembly
With the increasing impact of genomics in life sciences, the inference of high quality, reliable, and complete genome sequences is becoming critical. Genome assembly remains a major bottleneck in bioinformatics: indeed, high throughput sequencing apparatus yield millions of short sequencing reads that need to be merged based on their overlaps. Overlap graph based algorithms were used with the first generation of sequencers, while de Bruijn graph (DBG) based methods were preferred for the second generation. Because the sequencing coverage varies locally along the molecule, state-of-the-art assembly programs now follow an iterative process that requires the construction of de Bruijn graphs of distinct orders (i.e., sizes of the overlaps). The set of resulting sequences, termed unitigs, provide an important improvement compared to single DBG approaches. Here, we present a novel approach based on a digraph, the Superstring Graph, that captures all desired sizes of overlaps at once and allows to discard unreliable overlaps. With a simple algorithm, the Superstring Graph delivers sequences that includes all the unitigs obtained from multiple DBG as substrings. In linear time and space, it combines the efficiency of a greedy approach to the advantages of using a single graph. In summary, we present a first and formal comparison of the output of state-of-the-art genome assemblers.
YOC, A new strategy for pairwise alignment of collinear genomes
BACKGROUND:Comparing and aligning genomes is a key step in analyzing closely related genomes. Despite the development of many genome aligners in the last 15years, the problem is not yet fully resolved, even when aligning closely related bacterial genomes of the same species. In addition, no procedures are available to assess the quality of genome alignments or to compare genome aligners.RESULTS:We designed an original method for pairwise genome alignment, named YOC, which employs a highly sensitive similarity detection method together with a recent collinear chaining strategy that allows overlaps. YOC improves the reliability of collinear genome alignments, while preserving or even improving sensitivity. We also propose an original qualitative evaluation criterion for measuring the relevance of genome alignments. We used this criterion to compare and benchmark YOC with five recent genome aligners on large bacterial genome datasets, and showed it is suitable for identifying the specificities and the potential flaws of their underlying strategies.CONCLUSIONS:The YOC prototype is available at https://github.com/ruricaru/YOC webcite. It has several advantages over existing genome aligners: (1) it is based on a simplified two phase alignment strategy, (2) it is easy to parameterize, (3) it produces reliable genome alignments, which are easier to analyze and to use.
The ancient Yakuts: a population genetic enigma
This study is part of an ongoing project aiming at determining the ethnogenesis of an eastern Siberian ethnic group, the Yakuts, on the basis of archaeological excavations carried out over a period of 10 years in three regions of Yakutia: Central Yakutia, the Vilyuy River basin and the Verkhoyansk area. In this study, genetic analyses were carried out on skeletal remains from 130 individuals of unknown ancestry dated mainly from the fifteenth to the nineteenth century AD. Kinship studies were conducted using sets of commercially available autosomal and Y-chromosomal short tandem repeats (STRs) along with hypervariable region I sequences of the mitochondrial DNA. An unexpected and intriguing finding of this work was that the uniparental marker systems did not always corroborate results from autosomal DNA analyses; in some cases, false-positive relationships were observed. These discrepancies revealed that 15 autosomal STR loci are not sufficient to discriminate between first degree relatives and more distantly related individuals in our ancient Yakut sample. The Y-STR analyses led to similar conclusions, because the current Y-STR panels provided the limited resolution of the paternal lineages.
Construction of a de Bruijn Graph for Assembly from a Truncated Suffix Tree
In the life sciences, determining the sequence of bio-molecules is essential step towards the understanding of their functions and interactions inside an organism. Powerful technologies allows to get huge quantities of short sequencing reads that need to be assemble to infer the complete target sequence. These constraints favour the use of a version de Bruijn Graph (DBG) dedicated to assembly. The de Bruijn Graph is usually built directly from the reads, which is time and space consuming. Given a set $R$ of input words, well-known data structures, like the generalised suffix tree, can index all the substrings of words in $R$. In the context of DBG assembly, only substrings of length $k+1$ and some of length $k$ are useful. A truncated version of the suffix tree can index those efficiently. As indexes are exploited for numerous purposes in bioinformatics, as read cleaning, filtering, or even analysis, it is important to enable the community to reuse an existing index to build the DBG directly from it. In an earlier work we provided the first algorithms when starting from a suffix tree or suffix array. Here, we exhibit an algorithm that exploits a reduced version of the truncated suffix tree and computes the DBG from it. Importantly, a variation of this algorithm is also shown to compute the contracted DBG, which offers great benefits in practice. Both algorithms are linear in time and space in the size of the output.
Reverse engineering of compact suffix trees and links: A novel algorithm
Invented in the 1970s, the Suffix Tree (ST) is a data structure that indexes all substrings of a text in linear space. Although more space demanding than other indexes, the ST remains likely an inspiring index because it represents substrings in a hierarchical tree structure. Along time, STs have acquired a central position in text algorithmics with myriad of algorithms and applications to for instance motif discovery, biological sequence comparison, or text compression. It is well known that different words can lead to the same suffix tree structure with different labels. Moreover, the properties of STs prevent all tree structures from being STs. Even the suffix links, which play a key role in efficient construction algorithms and many applications, are not sufficient to discriminate the suffix trees of distinct words. The question of recognising which trees can be STs has been raised and termed Reverse Engineering on STs. For the case where a tree is given with potential suffix links, a seminal work provides a linear time solution only for binary alphabets. Here, we also investigate the Reverse Engineering problem on ST with links and exhibit a novel approach and algorithm. Hopefully, this new suffix tree characterisation makes up a valuable step towards a better understanding of suffix tree combinatorics.
LoRDEC: accurate and efficient long read error correction
Motivation: PacBio single molecule real-time sequencing is a third-generation sequencing technique producing long reads, with comparatively lower throughput and higher error rate. Errors include numerous indels and complicate downstream analysis like mapping or de novo assembly. A hybrid strategy that takes advantage of the high accuracy of second-generation short reads has been proposed for correcting long reads. Mapping of short reads on long reads provides sufficient coverage to eliminate up to 99% of errors, however, at the expense of prohibitive running times and considerable amounts of disk and memory space. Results : We present LoRDEC, a hybrid error correction method that builds a succinct de Bruijn graph representing the short reads, and seeks a corrective sequence for each erroneous region in the long reads by traversing chosen paths in the graph. In comparison, LoRDEC is at least six times faster and requires at least 93% less memory or disk space than available tools, while achieving comparable accuracy. Availability and implementaion : LoRDEC is written in C++, tested on Linux platforms and freely available at http://atgc.lirmm.fr/lordec . Contact:lordec@lirmm.fr
From Indexing Data Structures to de Bruijn Graphs
New technologies have tremendously increased sequencing throughput compared to traditional techniques, thereby complicating DNA assembly. Hence, assembly programs resort to de Bruijn graphs (dBG) of k-mers of short reads to compute a set of long contigs, each being a putative segment of the sequenced molecule. Other types of DNA sequence analysis, as well as preprocessing of the reads for assembly, use classical data structures to index all substrings of the reads. It is thus interesting to exhibit algorithms that directly build a dBG of order k from a pre-existing index, and especially a contracted version of the dBG, where non branching paths are condensed into single nodes. Here, we formalise the relationship between suffix trees/arrays and dBGs, and exhibit linear time algorithms for constructing the full or contracted dBGs. Finally, we provide hints explaining why this bridge between indexes and dBGs enables to dynamically update the order k of the graph.
Diversity of Prdm9 Zinc Finger Array in Wild Mice Unravels New Facets of the Evolutionary Turnover of this Coding Minisatellite
In humans and mice, meiotic recombination events cluster into narrow hotspots whose genomic positions are defined by the PRDM9 protein via its DNA binding domain constituted of an array of zinc fingers (ZnFs). High polymorphism and rapid divergence of the Prdm9 gene ZnF domain appear to involve positive selection at DNA-recognition amino-acid positions, but the nature of the underlying evolutionary pressures remains a puzzle. Here we explore the variability of the Prdm9 ZnF array in wild mice, and uncovered a high allelic diversity of both ZnF copy number and identity with the caracterization of 113 alleles. We analyze features of the diversity of ZnF identity which is mostly due to non-synonymous changes at codons −1, 3 and 6 of each ZnF, corresponding to amino-acids involved in DNA binding. Using methods adapted to the minisatellite structure of the ZnF array, we infer a phylogenetic tree of these alleles. We find the sister species Mus spicilegus and M. macedonicus as well as the three house mouse (Mus musculus) subspecies to be polyphyletic. However some sublineages have expanded independently in Mus musculus musculus and M. m. domesticus, the latter further showing phylogeographic substructure. Compared to random genomic regions and non-coding minisatellites, none of these patterns appears exceptional. In silico prediction of DNA binding sites for each allele, overlap of their alignments to the genome and relative coverage of the different families of interspersed repeated elements suggest a large diversity between PRDM9 variants with a potential for highly divergent distributions of recombination events in the genome with little correlation to evolutionary distance. By compiling PRDM9 ZnF protein sequences in Primates, Muridae and Equids, we find different diversity patterns among the three amino-acids most critical for the DNA-recognition function, suggesting different diversification timescales.
Approximation of Greedy Algorithms for Max-ATSP, Maximal Compression, Maximal Cycle Cover, and Shortest Cyclic Cover of Strings
Covering a directed graph by a Hamiltonian path or a set of words by a superstring belong to well studied optimisation problems that prove difficult to approximate. Indeed, the Maximum Asymmetric Travelling Salesman Problem (Max-ATSP), which asks for a Hamiltonian path of maximum weight covering a digraph, and the Shortest Superstring Problem (SSP), which, for a finite language P ≔ s_1, …, s_p, searches for a string of minimal length having each input word as a substring, are both Max-SNP hard. Finding a short superstring requires to choose a permutation of words and the associated overlaps to minimise the superstring length or to maximise the compression of P. Hence, a strong relation exists between Max-ATSP and SSP since solving Max-ATSP on the Overlap Graph for P gives a shortest superstring. Numerous works have designed algorithms that improve the approximation ratio but are increasingly complex. Often, these rely on solving the pendant problems where the cover is made of cycles instead of single path (Max-CC and SCCS). Finally, the greedy algorithm remains an attractive solution for its simplicity and ease of implementation. Its approximation ratios have been obtained by different approaches. In a seminal but complex proof, Tarhio and Ukkonen showed that it achieves 1/2 compression ratio for Max-CC. Here, using the full power of subset systems, we provide a unified approach for proving simply the approximation ratio of a greedy algorithm for these four problems. Especially, our proof for Maximal Compression shows that the Monge property suffices to derive the 1/2 tight bound.
An improved genome of the model marine alga Ostreococcus tauri unfolds by assessing Illumina de novo assemblies
BACKGROUND:Cost effective next generation sequencing technologies now enable the production of genomic datasets for many novel planktonic eukaryotes, representing an understudied reservoir of genetic diversity. O. tauri is the smallest free-living photosynthetic eukaryote known to date, a coccoid green alga that was first isolated in 1995 in a lagoon by the Mediterranean sea. Its simple features, ease of culture and the sequencing of its 13Mb haploid nuclear genome have promoted this microalga as a new model organism for cell biology. Here, we investigated the quality of genome assemblies of Illumina GAIIx 75bp paired-end reads from Ostreococcus tauri, thereby also improving the existing assembly and showing the genome to be stably maintained in culture.RESULTS:The 3 assemblers used, ABySS, CLCBio and Velvet, produced 95% complete genomes in 1402 to 2080 scaffolds with a very low rate of misassembly. Reciprocally, these assemblies improved the original genome assembly by filling in 930 gaps. Combined with additional analysis of raw reads and PCR sequencing effort, 1194 gaps have been solved in total adding up to 460kb of sequence. Mapping of RNAseq Illumina data on this updated genome led to a twofold reduction in the proportion of multi-exon protein coding genes, representing 19% of the total 7699 protein coding genes. The comparison of the DNA extracted in 2001 and 2009 revealed the fixation of 8 single nucleotide substitutions and 2 deletions during the approximately 6000 generations in the lab. The deletions either knocked out or truncated two predicted transmembrane proteins, including a glutamate-receptor like gene.CONCLUSION:High coverage (>80 fold) paired-end Illumina sequencing enables a high quality 95% complete genome assembly of a compact ~13Mb haploid eukaryote. This genome sequence has remained stable for 6000 generations of lab culture.
Scalable and Versatile k-mer Indexing for High-Throughput Sequencing Data
Philippe et al. (2011) proposed a data structure called Gk arrays for indexing and querying large collections of high-throughput sequencing data in main-memory. The data structure supports versatile queries for counting, locating, and analysing the coverage profile of k-mers in short-read data. The main drawback of the Gk arrays is its space-consumption, which can easily reach tens of gigabytes of main-memory even for moderate size inputs. We propose a compressed variant of Gk arrays that supports the same set of queries, but in both near-optimal time and space. In practice, the compressed Gk arrays scale up to much larger inputs with highly competitive query times compared to its non-compressed predecessor. The main applications include variant calling, error correction, coverage profiling, and sequence assembly.
CRAC: an integrated approach to the analysis of RNA-seq reads
A large number of RNA-sequencing studies set out to predict mutations, splice junctions or fusion RNAs. We propose a method, CRAC, that integrates genomic locations and local coverage to enable such predictions to be made directly from RNA-seq read analysis. A k-mer profiling approach detects candidate mutations, indels and splice or chimeric junctions in each single read. CRAC increases precision compared with existing tools, reaching 99:5% for splice junctions, without losing sensitivity. Importantly, CRAC predictions improve with read length. In cancer libraries, CRAC recovered 74% of validated fusion RNAs and predicted novel recurrent chimeric junctions. CRAC is available at http://crac.gforge.inria.fr.
Combining DGE and RNA-sequencing data to identify new polyA+ non-coding transcripts in the human genome
Recent sequencing technologies that allow massive parallel production of short reads are the method of choice for transcriptome analysis. Particularly, digital gene expression (DGE) technologies produce a large dynamic range of expression data by generating short tag signatures for each cell transcript. These tags can be mapped back to a reference genome to identify new transcribed regions that can be further covered by RNA-sequencing (RNA-Seq) reads. Here, we applied an integrated bioinformatics approach that combines DGE tags, RNA-Seq, tiling array expression data and species-comparison to explore new transcriptional regions and their specific biological features, particularly tissue expression or conservation. We analysed tags from a large DGE data set (designated as TranscriRef). We then annotated 750 000 tags that were uniquely mapped to the human genome according to Ensembl. We retained transcripts originating from both DNA strands and categorized tags corresponding to protein-coding genes, antisense, intronic- or intergenic-transcribed regions and computed their overlap with annotated non-coding transcripts. Using this bioinformatics approach, we identified ~34 000 novel transcribed regions located outside the boundaries of known protein-coding genes. As demonstrated using sequencing data from human pluripotent stem cells for biological validation, the method could be easily applied for the selection of tissue-specific candidate transcripts. DigitagCT is available at http://cractools.gforge.inria.fr/softwares/digitagct.
Approximate Common Intervals in Multiple Genome Comparison
We consider the problem of inferring approximate common intervals of multiple genomes. Genomes are modelled as sequences of homologous genes families identifiers, and approximate common intervals represent conserved regions possibly showing rearrangements, as well as repetitions, or insertions/deletions. This problem is already known, but existing approaches are not incremental and somehow limited to special cases. We adopt a simple, classical graph-based approach, where the vertices of the graph represent the exact common intervals of the sequences (i.e., regions containing the same gene set), and where edges link vertices that differ by less than δ elements (with δ being parameter). With this model, approximate gene clusters are maximal cliques of the graph: computing them can exploit known and well designed algorithms. For a proof of concept, we applied the method to several datasets of bacterial genomes and compared the two maximal cliques algorithms, a static and a dynamic one. While being quite flexible, this approach opens the way to a combinatorial characterization of genomic rearrangements in terms of graph substructures.
Novel Definition and Algorithm for Chaining Fragments with Proportional Overlaps
Abstract Chaining fragments is a crucial step in genome alignment. Existing chaining algorithms compute a maximum weighted chain with no overlaps allowed between adjacent fragments. In practice, using local alignments as fragments, instead of Maximal Exact Matches (MEMs), generates frequent overlaps between fragments, due to combinatorial reasons and biological factors, i.e., variable tandem repeat structures that differ in number of copies between genomic sequences. In this article, in order to raise this limitation, we formulate a novel definition of a chain, allowing overlaps proportional to the fragments lengths, and exhibit an efficient algorithm for computing such a maximum weighted chain. We tested our algorithm on a dataset composed of 694 genome pairs and accounted for significant improvements in terms of coverage, while keeping the running times below reasonable limits. Moreover, experiments with different ratios of allowed overlaps showed the robustness of the chains with respect to these ratios. Our algorithm is implemented in a tool called OverlapChainer (OC), which is available upon request to the authors.
Evolutionary process of a tetranucleotide microsatellite locus in Acipenseriforms
The evolutionary dynamics of the tetra-nucleotide microsatellite locus Spl-106 were investigated at the repeat and flanking sequences in 137 individuals of 15 Acipenseriform species, giving 93 homologous sequences, which were detected in 11 out of 15 species. Twenty-three haplotypes of flanking sequences and three distinct types of repeats, type I, type II and type III, were found within these 93 sequences. The MS-Align phylogenetic method, newly applied to microsatellite sequences, permitted us to understand the repeat and flanking sequence evolution of Spl-106 locus. The flanking region of locus Spl-106 was highly conserved among the species of genera Acipenser, Huso and Scaphirhynchus, which diverged about 150 million years ago (Mya). The rate of flanking sequence divergence at the microsatellite locus Spl-106 in sturgeons is between 0.011% and 0.079% with an average at 0.028% per million years. Sequence alignment and phylogenetic trees produced by MS-Align showed that both the flanking and repeat regions can cluster the alleles of different species into Pacific and Atlantic lineages. Our results show a synchronous evolutionary pattern between the flanking and repeat regions. Moreover, the coexistence of different repeat types in the same species, even in the same individual, is probably due to two duplication events encompassing the locus Spl-106 that occurred during the divergence of Pacific lineage. The first occured before the diversification of Pacific species (121-96 Mya) and led to repeat types I and II. The second occurred more recently, just before the speciation of A. sinensis and A. dabryanus (69-10 Mya), and led to repeat type III. Sequences in the same species with different repeat types probably corresponds to paralogous loci. This study sheds a new light on the evolutionary mechanisms that shape the complex microsatellite loci involving different repeat types.
An alternative approach to multiple genome comparison
Genome comparison is now a crucial step for genome annotation and identification of regulatory motifs. Genome comparison aims for instance at finding genomic regions either specific to or in one-to-one correspondance between individuals/strains/species. It serves e.g. to pre-annotate a new genome by automatically transfering annotations from a known one. However, efficiency, flexibility and objectives of current methods do not suit the whole spectrum of applications, genome sizes and organizations. Innovative approaches are still needed. Hence, we propose an alternative way of comparing multiple genomes based on segmentation by similarity. In this framework, rather than being formulated as a complex optimization problem, genome comparison is seen as a segmentation question for which a single optimal solution can be found in almost linear time. We apply our method to analyse three strains of a virulent pathogenic bacteria, Ehrlichia ruminantium, and identify 92 new genes. We also find out that a substantial number of genes thought to be strain specific have potential orthologs in the other strains. Our solution is implemented in an efficient program, qod, equipped with a user-friendly interface, and enables the automatic transfer of annotations betwen compared genomes or contigs (Video in Supplementary Data). Because it somehow disregards the relative order of genomic blocks, qod can handle unfinished genomes, which due to the difficulty of sequencing completion may become an interesting characteristic for the future. Availabilty: http://www.atgc-montpellier.fr/qod.
Querying large read collections in main memory: a versatile data structure
BACKGROUND:High Throughput Sequencing (HTS) is now heavily exploited for genome (re-)sequencing, metagenomics, epigenomics, and transcriptomics and requires different, but computer intensive bioinformatic analyses. When a reference genome is available, mapping reads on it is the first step of this analysis. Read mapping programs owe their efficiency to the use of involved genome indexing data structures, like the Burrows-Wheeler transform. Recent solutions index both the genome, and the k-mers of the reads using hash-tables to further increase efficiency and accuracy. In various contexts (e.g. assembly or transcriptome analysis), read processing requires to determine the sub-collection of reads that are related to a given sequence, which is done by searching for some k-mers in the reads. Currently, many developments have focused on genome indexing structures for read mapping, but the question of read indexing remains broadly unexplored. However, the increase in sequence throughput urges for new algorithmic solutions to query large read collections efficiently. RESULTS:Here, we present a solution, named Gk arrays, to index large collections of reads, an algorithm to build the structure, and procedures to query it. Once constructed, the index structure is kept in main memory and is repeatedly accessed to answer queries like “given a k-mer, get the reads containing this k-mer (once/at least once)". We compared our structure to other solutions that adapt uncompressed indexing structures designed for long texts and show that it processes queries fast, while requiring much less memory. Our structure can thus handle larger read collections. We provide examples where such queries are adapted to different types of read analysis (SNP detection, assembly, RNA-Seq). CONCLUSIONS:Gk arrays constitute a versatile data structure that enables fast and more accurate read analysis in various contexts. The Gk arrays provide a flexible brick to design innovative programs that mine efficiently genomics, epigenomics, metagenomics, or transcriptomics reads. The Gk arrays library is available under Cecill (GPL compliant) license from http://www.atgc-montpellier.fr/ngs/.
Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features
In metazoans, thousands of DNA replication origins (Oris) are activated at each cell cycle. Their genomic organization and their genetic nature remain elusive. Here, we characterized Oris by nascent strand (NS) purification and a genome-wide analysis in Drosophila and mouse cells. We show that in both species most CpG islands (CGI) contain Oris, although methylation is nearly absent in Drosophila, indicating that this epigenetic mark is not crucial for defining the activated origin. Initiation of DNA synthesis starts at the borders of CGI, resulting in a striking bimodal distribution of NS, suggestive of a dual initiation event. Oris contain a unique nucleotide skew around NS peaks, characterized by G/T and C/A overrepresentation at the 5’ and 3’ of Ori sites, respectively. Repeated GC-rich elements were detected, which are good predictors of Oris, suggesting that common sequence features are part of metazoan Oris. In the heterochromatic chromosome 4 of Drosophila, Oris correlated with HP1 binding sites. At the chromosome level, regions rich in Oris are early replicating, whereas Ori-poor regions are late replicating. The genome-wide analysis was coupled with a DNA combing analysis to unravel the organization of Oris. The results indicate that Oris are in a large excess, but their activation does not occur at random. They are organized in groups of site-specific but flexible origins that define replicons, where a single origin is activated in each replicon. This organization provides both site specificity and Ori firing flexibility in each replicon, allowing possible adaptation to environmental cues and cell fates.
Novel definition and algorithm for chaining fragments with proportional overlaps
Chaining fragments is a crucial step in genome alignment. Existing chaining algorithms compute a maximum weighted chain with no overlaps allowed between adjacent fragments. In practice, using local alignments as fragments, instead of MEMs, generates frequent overlaps between fragments, due to combinatorial reasons and biological factors, i.e. variable tandem repeat structures that differ in number of copies between genomic sequences. In this paper, in order to raise this limitation, we formulate a novel definition of a chain, allowing overlaps proportional to the fragments lengths, and exhibit an efficient algorithm for computing such a maximum weighted chain. We tested our algorithm on a dataset composed of 694 genome couples and accounted for significant improvements in terms of coverage, while keeping the running times below reasonable limits.
DNA Slippage Occurs at Microsatellite Loci without Minimal Threshold Length in Humans: A Comparative Genomic Approach
The dynamics of microsatellite, or short tandem repeats (STRs), is well documented for long, polymorphic loci, but much less is known for shorter ones. For example, the issue of a minimum threshold length for DNA slippage remains contentious. Model-fitting methods have generally concluded that slippage only occurs over a threshold length of about eight nucleotides, in contradiction with some direct observations of tandem duplications at shorter repeated sites. Using a comparative analysis of the human and chimpanzee genomes, we examined the mutation patterns at microsatellite loci with lengths as short as one period plus one nucleotide. We found that the rates of tandem insertions and deletions at microsatellite loci strongly deviated from background rates in other parts of the human genome and followed an exponential increase with STR size. More importantly, we detected no lower threshold length for slippage. The rate of tandem duplications at unrepeated sites was higher than expected from random insertions, providing evidence for genome-wide action of indel slippage (an alternative mechanism generating tandem repeats). The rate of point mutations adjacent to STRs did not differ from that estimated elsewhere in the genome, except around dinucleotide loci. Our results suggest that the emergence of STR depends on DNA slippage, indel slippage, and point mutations. We also found that the dynamics of tandem insertions and deletions differed in both rates and size at which these mutations take place. We discuss these results in both evolutionary and mechanistic terms.
Sequence analysis of two alleles reveals that intra-and intergenic recombination played a role in the evolution of the radish fertility restorer (Rfo)
Background: Land plant genomes contain multiple members of a eukaryote-specific gene family encoding proteins with pentatricopeptide repeat (PPR) motifs. Some PPR proteins were shown to participate in post-transcriptional events involved in organellar gene expression, and this type of function is now thought to be their main biological role. Among PPR genes, restorers of fertility (Rf) of cytoplasmic male sterility systems constitute a peculiar subgroup that is thought to evolve in response to the presence of mitochondrial sterility-inducing genes. Rf genes encoding PPR proteins are associated with very close relatives on complex loci. Results: We sequenced a non-restoring allele (L7rfo) of the Rfo radish locus whose restoring allele (D81Rfo) was previously described, and compared the two alleles and their PPR genes. We identified a ca 13 kb long fragment, likely originating from another part of the radish genome, inserted into the L7rfo sequence. The L7rfo allele carries two genes (PPR-1 and PPR-2) closely related to the three previously described PPR genes of the restorer D81Rfo allele (PPR-A, PPR-B, and PPR-C). Our results indicate that alleles of the Rfo locus have experienced complex evolutionary events, including recombination and insertion of extra-locus sequences, since they diverged. Our analyses strongly suggest that present coding sequences of Rfo PPR genes result from intragenic recombination. We found that the 10 C-terminal PPR repeats in Rfo PPR gene encoded proteins result from the tandem duplication of a 5 PPR repeat block. Conclusions: The Rfo locus appears to experience more complex evolution than its flanking sequences. The Rfo locus and PPR genes therein are likely to evolve as a result of intergenic and intragenic recombination. It is therefore not possible to determine which genes on the two alleles are direct orthologs. Our observations recall some previously reported data on pathogen resistance complex loci.
MPSCAN: Fast Localisation of Multiple Reads in Genomes
With Next Generation Sequencers, sequence based transcriptomic or epigenomic assays yield millions of short sequence reads that need to be mapped back on a reference genome. The upcoming versions of these sequencers promise even higher sequencing capacities; this may turn the read mapping task into a bottleneck for which alternative pattern matching approaches must be experimented. We present an algorithm and its implementation, called mpscan, which uses a sophisticated filtration scheme to match a set of patterns/reads exactly on a sequence. mpscan can search for millions of reads in a single pass through the genome without indexing its sequence. Moreover, we show that mpscan offers an optimal average time complexity, which is sublinear in the text length, meaning that it does not need to examine all sequence positions. Comparisons with BLAT-like tools and with six specialised read mapping programs (like bowtie or zoom) demonstrate that mpscan also is the fastest algorithm in practice for exact matching. Our accuracy and scalability comparisons reveal that some tools are inappropriate for read mapping. Moreover, we provide evidence suggesting that exact matching may be a valuable solution in some read mapping applications. As most read mapping programs somehow rely on exact matching procedures to perform approximate pattern mapping, the filtration scheme we experimented may reveal useful in the design of future algorithms. The absence of genome index gives mpscan its low memory requirement and flexibility that let it run on a desktop computer and avoids a time-consuming genome preprocessing.
Using reads to annotate the genome: influence of length, background distribution, and sequence errors on prediction capacity
Ultra high-throughput sequencing is used to analyse the transcriptome or interactome at unprecedented depth on a genome-wide scale. These techniques yield short sequence reads that are then mapped on a genome sequence to predict putatively transcribed or protein-interacting regions. We argue that factors such as background distribution, sequence errors, and read length impact on the prediction capacity of sequence census experiments. Here we suggest a computational approach to measure these factors and analyse their influence on both transcriptomic and epigenomic assays. This investigation provides new clues on both methodological and biological issues. For instance, by analysing chromatin immunoprecipitation read sets, we estimate that 4.6% of reads are affected by SNPs. We show that, although the nucleotide error probability is low, it significantly increases with the position in the sequence. Choosing a read length above 19 bp practically eliminates the risk of finding irrelevant positions, while above 20 bp the number of uniquely mapped reads decreases. With our procedure, we obtain 0.6% false positives among genomic locations. Hence, even rare signatures should identify biologically relevant regions, if they are mapped on the genome. This indicates that digital transcriptomics may help to characterize the wealth of yet undiscovered, low-abundance transcripts.
Hardness of optimal spaced seed design
Speeding up approximate pattern matching is a line of research in stringology since the 80s. Practically fast approaches belong to the class of filtration algorithms, in which text regions dissimilar to the pattern are first excluded, and the remaining regions are then compared to the pattern by dynamic programming. Among the conditions used to test similarity between the regions and the pattern, many require a minimum number of common substrings between them. When only substitutions are taken into account for measuring dissimilarity, counting spaced subwords instead of substrings improves the filtration efficiency. However, a preprocessing step is required to design one or more patterns, called spaced seeds (or gapped seeds), for the subwords, depending on the search parameters. Two distinct lines of research appear the literature: one with probabilistic formulations of seed design problems, in which one wishes for instance to compute a seed with the highest probability to detect the desired similarities (lossy filtration), a second line with combinatorial formulations, where the goal is to find a seed that detects all or a maximum number of similarities (both lossless and lossy filtration). We concentrate on combinatorial seed design problems and consider formulations in which the set of sought similarities is either listed explicitly (RSOS), or characterised by their length and maximal number of mismatches (Non-Detection). Several articles exhibit exponential algorithms for these problems. In this work, we provide hardness and inapproximability results for several seed design problems, thereby justifying the complexity of known algorithms. Moreover, we introduce a new formulation of seed design (MWLS), in which the weight of the seed has to be maximised, and show it is as difficult to approximate as Maximum Independent Set.
Detection of Recombination in Variable Number Tandem Repeat Sequences
Tandem repeats are repeated sequences whose copies are adjacent along the chromosomes. They account for large portion of eukaryotic genomes and are found in all types of living organisms. Among tandem repeats, those with repeat unit of middle size are called minisatellites. These loci depart from classical loci because of the propensity to vary in size due to the addition or the removal of one or more repeat units. Due to this polymorphism, they prove useful in genetic mapping, in population genetics, and forensic medicine. Moreover, some specific tandem repeat loci are involved in diseases, like the insulin minisatellite, which is implicated in type I diabetes and obesity. Those loci also undergo complex recombination events. Presently, some programs to compare tandem repeats alleles exist and yield good results when recombination is absent, but none correctly handles recombinant alleles. Our goal is to develop an adequate tool for the detection of recombinant among a set of minisatellite sequences. By combining a multiple alignment tool and a method based on phylogenetic profiling, we design a first solution, called MS_PhylPro, for this task. The method has been implemented, tested on real data sets from the insulin minisatellite, and proven to detect recombinant alleles.
Transcriptome annotation using tandem SAGE tags
Analysis of several million expressed gene signatures (tags) revealed an increasing number of different sequences, largely exceeding that of annotated genes in mammalian genomes. Serial analysis of gene expression (SAGE) can reveal new Poly(A) RNAs transcribed from previously unrecognized chromosomal regions. However, conventional SAGE tags are too short to identify unambiguously unique sites in large genomes. Here, we design a novel strategy with tags anchored on two different restrictions sites of cDNAs. New transcripts are then tentatively defined by the two SAGE tags in tandem and by the spanning sequence read on the genome between these tagged sites. Having developed a new algorithm to locate these tag-delimited genomic sequences (TDGS), we first validated its capacity to recognize known genes and its ability to reveal new transcripts with two SAGE libraries built in parallel from a single RNA sample. Our algorithm proves fast enough to experiment this strategy at a large scale. We then collected and processed the complete sets of human SAGE tags to predict yet unknown transcripts. A cross-validation with tiling arrays data shows that 47% of these TDGS overlap transcriptional active regions. Our method provides a new and complementary approach for complex transcriptome annotation.
Detecting microsatellites within genomes: significant variation among algorithms
Background: Microsatellites are short, tandemly-repeated DNA sequences which are widely distributed among genomes. Their structure, role and evolution can be analyzed based on exhaustive extraction from sequenced genomes. Several dedicated algorithms have been developed for this purpose. Here, we compared the detection efficiency of five of them (TRF, Mreps, Sputnik, STAR, and RepeatMasker). Results: Our analysis was first conducted on the human X chromosome, and microsatellite distributions were characterized by microsatellite number, length, and divergence from a pure motif. The algorithms work with user-defined parameters, and we demonstrate that the parameter values chosen can strongly influence microsatellite distributions. The five algorithms were then compared by fixing parameters settings, and the analysis was extended to three other genomes (Saccharomyces cerevisiae, Neurospora crassa and Drosophila melanogaster) spanning a wide range of size and structure. Significant differences for all characteristics of microsatellites were observed among algorithms, but not among genomes, for both perfect and imperfect microsatellites. Striking differences were detected for short microsatellites (below 20 bp), regardless of motif. Conclusion: Since the algorithm used strongly influences empirical distributions, studies analyzing microsatellite evolution based on a comparison between empirical and theoretical size distributions should therefore be considered with caution. We also discuss why a typological definition of microsatellites limits our capacity to capture their genomic distributions.
Longest common subsequence problem for unoriented and cyclic strings
Given a finite set of strings X, the Longest Common Subsequence problem (LCS) consists in finding a subsequence common to all strings in X that is of maximal length. LCS is a central problem in stringology and finds broad applications in text compression, conception of error-detecting codes, or biological sequence comparison. However, in numerous contexts, words represent cyclic or unoriented sequences of symbols and LCS must be generalized to consider both orientations and/or all cyclic shifts of the strings involved. This occurs especially in computational biology when genetic material is sequenced from circular DNA or RNA molecules. In this work, we define three variants of LCS when the input words are unoriented and/or cyclic. We show that these problems are NP-hard, and W[1]-hard if parameterized in the number of input strings. These results still hold even if the three LCS variants are restricted to input languages over a binary alphabet. We also settle the parameterized complexity of our problems for most relevant parameters. Moreover, we study the approximability of these problems: we discuss the existence of approximation bounds depending on the cardinality of the alphabet, on the length of the shortest sequence, and on the number of input sequences. For this we prove that Maximum Independent Set in r-uniform hypergraphs is W[1]-hard if parameterized in the cardinality of the sought independent set and at least as hard to approximate as Maximum Independent Set in graphs.
Species-wide distribution of highly polymorphic minisatellite markers suggests past and present genetic exchanges among house mouse subspecies
Background: Four hypervariable minisatellite loci were scored on a panel of 116 individuals of various geographical origins representing a large part of the diversity present in house mouse subspecies. Internal structures of alleles were determined by minisatellite variant repeat mapping PCR to produce maps of intermingled patterns of variant repeats along the repeat array. To reconstruct the genealogy of these arrays of variable length, the specifically designed software MS_Align was used to estimate molecular divergences, graphically represented as neighbor-joining trees. Results: Given the high haplotypic diversity detected (mean He = 0.962), these minisatellite trees proved to be highly informative for tracing past and present genetic exchanges. Examples of identical or nearly identical alleles were found across subspecies and in geographically very distant locations, together with poor lineage sorting among subspecies except for the X-chromosome locus MMS30 in Mus mus musculus. Given the high mutation rate of mouse minisatellite loci, this picture cannot be interpreted only with simple splitting events followed by retention of polymorphism, but implies recurrent gene flow between already differentiated entities. Conclusion: This strongly suggests that, at least for the chromosomal regions under scrutiny, wild house mouse subspecies constitute a set of interrelated gene pools still connected through long range gene flow or genetic exchanges occurring in the various contact zones existing nowadays or that have existed in the past. Identifying genomic regions that do not follow this pattern will be a challenging task for pinpointing genes important for speciation.
Formation of the Arabidopsis Pentatricopeptide Repeat Family
In Arabidopsis (Arabidopsis thaliana) the 466 pentatricopeptide repeat (PPR) proteins are putative RNA-binding proteins with essential roles in organelles. Roughly half of the PPR proteins form the plant combinatorial and modular protein (PCMP) subfamily, which is land-plant specific. PCMPs exhibit a large and variable tandem repeat of a standard pattern of three PPR variant motifs. The association or not of this repeat with three non-PPR motifs at their C terminus defines four distinct classes of PCMPs. The highly structured arrangement of these motifs and the similar repartition of these arrangements in the four classes suggest precise relationships between motif organization and substrate specificity. This study is an attempt to reconstruct an evolutionary scenario of the PCMP family. We developed an innovative approach based on comparisons of the proteins at two levels: namely the succession of motifs along the protein and the amino acid sequence of the motifs. It enabled us to infer evolutionary relationships between proteins as well as between the inter- and intraprotein repeats. First, we observed a polarized elongation of the repeat from the C terminus toward the N-terminal region, suggesting local recombinations of motifs. Second, the most N-terminal PPR triple motif proved to evolve under different constraints than the remaining repeat. Altogether, the evidence indicates different evolution for the PPR region and the C-terminal one in PCMPs, which points to distinct functions for these regions. Moreover, local sequence homogeneity observed across PCMP classes may be due to interclass shuffling of motifs, or to deletions/insertions of non-PPR motifs at the C terminus.
Tiling an Interval of the Discrete Line
We consider the problem of tiling a segment [0,n] of the discrete line. More precisely, we ought to characterize the structure of the patterns that tile a segment and their number. A pattern is a subset of $N$. A tiling pattern or tile for [0,n] is a subset $A ın a̧l P(N)$ such that there exists $B ın ļ P(N)$ and such that the direct sum of $A$ and $B$ equals [0,n]. This problem is related to the difficult question of the decomposition in direct sums of the torus $Z/nZ$ (proposed by Minkowski). Using combinatorial and algebraic techniques, we give a new elementary proof of Krasner factorizations. We combinatorially prove that the tiles are direct sums of some arithmetic sequences of specific lengths. Besides, we show there are as many tiles whose smallest tilable segment is [0,n] as tiles whose smallest tilable segment is [0, d], for all strict divisors $d$ of $n$. This enables us to exhibit an optimal linear time algorithm to compute for a given pattern the smallest segment that it tiles if any, as well as a recurrence formula for counting the tiles of a segment.
A Fast and Specific Alignment Method for Minisatellite Maps
Background Variable minisatellites count among the most polymorphic markers of eukaryotic and prokaryotic genomes. This variability can affect gene coding regions, like in the prion protein gene, or gene regulation regions, like for the cystatin B gene, and be associated or implicated in diseases: the Creutzfeld-Jakob disease and the myoclonus epilepsy type 1, for our examples. When it affects neutrally evolving regions, the polymorphism in length (i.e., in number of copies) of minisatellites proved useful in population genetics. Motivation In these tandem repeat sequences, different mutational mechanisms let the number of copies, as well as the copies themselves, vary. Especially, the interspersion of events of tandem duplication/contraction and of punctual mutation makes the succession of variant repeats much more informative than the sole allele length. To exploit this information requires the ability to align minisatellite alleles by accounting for both punctual mutations and tandem duplications. Results We propose a minisatellite maps alignment program that improves on previous solutions. Our new program is faster, simpler, considers an extended evolutionary model, and is available to the community. We test it on the data set of 609 alleles of the MSY1 (DYF155S1) human minisatellite and confirm its ability to recover known evolutionary signals. Our experiments highlight that the informativeness of minisatellites resides in their length and composition polymorphisms. Exploiting both simultaneously is critical to unravel the implications of variable minisatellites in the control of gene expression and diseases. Availability: Software is available at http://atgc.lirmm.fr/ms_align/
Hardness results for the center and median string problems under the weighted and unweighted edit distances
Given a finite set of strings, the Median String problem consists in finding a string that minimizes the sum of the edit distances to the strings in the set. Approximations of the median string are used in a very broad range of applications where one needs a representative string that summarizes common information to the strings of the set. It is the case in classification, in speech and pattern recognition, and in computational biology. In the latter, Median String is related to the key problem of multiple alignment. In the recent literature, one finds a theorem stating the NP-completeness of the Median String for unbounded alphabets. However, in the above mentioned areas, the alphabet is often finite. Thus, it remains a crucial question whether the Median String problem is NP-complete for bounded and even binary alphabets. In this work, we provide an answer to this question and also give the complexity of the related Center String problem. Moreover, we study the parameterized complexity of both problems with respect to the number of input strings. In addition, we provide an algorithm to compute an optimal center under a weighted edit distance in polynomial time when the number of input strings is fixed.
Hardness of Optimal Spaced Seed Design
Speeding up approximate pattern matching is a line of research in stringology since the 80’s. Practically fast approaches belong to the class of filtration algorithms, in which text regions dissimilar to the pattern are excluded (filtered out) in a first step, and remaining regions are compared to the pattern by dynamic programming in a second step. Among the necessary conditions used to test similarity between the regions and the pattern, many require a minimum number of common substrings between them. When only substitutions are taken into account for measuring dissimilarity, it was shown recently that counting spaced subwords instead of substrings improve the filtration efficiency. However, a preprocessing step is required to design one or more patterns, called gapped seeds, for the subwords, depending on the search parameters. The seed design problems proposed up to now differ by the way the similarities to detect are given: either a set of similarities is given in extenso (this is a “region specific” problem), or one wishes to detect all similar regions having at most k substitutions (general detection problem). Several articles exhibit exponential algorithms for these problems. In this work, we provide hardness and inapproximability results for both the region specific and general seed design problems, thereby justifying the exponential complexity of known algorithms. Moreover, we introduce a new formulation of the region specific seed design problem, in which the weight of the seed (i.e., number of characters in the subwords) has to be maximized, and show it is as difficult to approximate than Maximum Independent Set.
STAR: an algorithm to Search for Tandem Approximate Repeats
Motivation: Tandem repeats consist in approximate and adjacent repetitions of a DNA motif. Such repeats account for large portions of eukaryotic genomes and have also been found in other life kingdoms. Owing to their polymorphism, tandem repeats have proven useful in genome cartography, forensic and population studies, etc. Nevertheless, they are not systematically detected nor annotated in genome projects. Partially because of this lack of data, their evolution is still poorly understood. Results: In this work, we design an exact algorithm to locate approximate tandem repeats (ATR) of a motif in a DNA sequence. Given a motif and a DNA sequence, our method named STAR, identifies all segments of the sequence that correspond to significant approximate tandem repetitions of the motif. In our model, an Exact Tandem Repeat (ETR) comes from the tandem duplication of the motif and an ATR derives from an ETR by a series of point mutations. An ATR can then be encoded as a number of duplications of the motif together with a list of mutations. Consequently, any sequence that is not an ATR cannot be encoded efficiently by this description, while a true ATR can. Our method uses the minimum description length criterion to identify which sequence segments are ATR. Our optimization procedure guarantees that STAR finds a combination of ATR that minimizes this criterion. Availability: for use at http://atgc.lirmm.fr/star Supplementary information: an appendix is available at http://atgc.lirmm.fr/star under ‘Paper and contacts’.
A survey on algorithmic aspects of tandem repeats evolution
Local repetitions in genomes are called tandem repeats. A tandem repeat contains multiple, but slightly different copies of a repeated unit. It changes over time as the copies are altered by mutations, when additional copies are created by amplification of an existing copy, or when a copy is removed by contraction. Theses changes let tandem repeats evolve dynamically. From this statement follow two problems. TANDEM REPEAT HISTORY aims at recovering the history of amplifications and mutations that produced the tandem repeat sequence given as input. Given the tandem repeat sequences at the same genomic location in two individuals and a cost function for amplifications, contractions, and mutations, the purpose of TANDEM REPEAT ALLELE ALIGNMENT is to find an alignment of the sequences having minimal cost. We present a survey of these two problems that allow to investigate evolutionary mechanisms at work in tandem repeats.
Combinatorics of periods in strings
We consider the set Γn of all period sets of strings of length n over a finite alphabet. We show that there is redundancy in period sets and introduce the notion of an irreducible period set. We prove that Γn is a lattice under set inclusion and does not satisfy the Jordan–Dedekind condition. We propose the first efficient enumeration algorithm for Γn and improve upon the previously known asymptotic lower bounds on the cardinality of Γn. Finally, we provide a new recurrence to compute the number of strings sharing a given period set, and exhibit an algorithm to sample uniformly period sets through irreducible period set.
Comparison of Minisatellites
In the class of repeated sequences that occur in DNA, minisatellites have been found polymorphic and became useful tools in genetic mapping and forensic studies. They consist of a heterogeneous tandem array of a short repeat unit. The slightly different units along the array are called variants. Minisatellites evolve mainly through tandem duplications and tandem deletions of variants. Jeffreys et al. (1997) devised a method to obtain the sequence of variants along the array in a digital code and called such sequences maps. Minisatellite maps give access to the detail of mutation processes at work on such loci. In this paper, we design an algorithm to compare two maps under an evolutionary model that includes deletion, insertion, mutation, tandem duplication, and tandem deletion of a variant. Our method computes an optimal alignment in reasonable time; and the alignment score, i.e., the weighted sum of its elementary operations, is a distance metric between maps. The main difficulty is that the optimal sequence of operations depends on the order in which they are applied to the map. Taking the maps of the minisatellite MSY1 of 609 men, we computed all pairwise distances and reconstructed an evolutionary tree of these individuals. MSY1 (DYF155S1) is a hypervariable locus on the Y chromosome. In our tree, the populations of some haplogroups are monophyletic, showing that one can decipher a microevolutionary signal using minisatellite maps comparison.
On the Distribution of the Number of Missing Words in Random Texts
Determining the distribution of the number of empty urns after a number of balls have been thrown randomly into the urns is a classical and well understood problem. We study a generalization: Given a finite alphabet of size σ and a word length q, what is the distribution of the number X of words (of length q) that do not occur in a random text of length n+q−1 over the given alphabet? For q=1, X is the number Y of empty urns with σ urns and n balls. For q=2, X is related to the number Y of empty urns with σq urns and n balls, but the law of X is more complicated because successive words in the text overlap. We show that, perhaps surprisingly, the laws of X and Y are not as different as one might expect, but some problems remain currently open.
Complexities of the Centre and Median String Problems
Given a finite set of strings, the median string problem consists in finding a string that minimizes the sum of the distances to the strings in the set. Approximations of the median string are used in a very broad range of applications where one needs a representative string that summarizes common information to the strings of the set. It is the case in Classification, in Speech and Pattern Recognition, and in Computational Biology. In the latter, Median String is related to the key problem of Multiple Alignment. In the recent literature, one finds a theorem stating the NP-completeness of the median string for unbounded alphabets. However, in the above mentioned areas, the alphabet is often finite. Thus, it remains a crucial question whether the median string problem is NP-complete for finite and even binary alphabets. In this work, we provide an answer to this question and also give the complexity of the related centre string problem. Moreover, we study the parametrized complexity of both problems with respect to the number of input strings.
Comparison of minisatellites
In the class of repeated sequences that occur in DNA, minisatellites have been found polymorphic and became useful tools in genetic mapping and forensic studies. They consist of a heterogeneous tandem array of a short repeat unit. The slightly different units along the array are called variants. Minisatellites evolve mainly through tandem duplications and tandem deletions of variants. Jeffreys et al. devised a method to obtain the sequence of variants along the array in a digital code, and called such sequences maps. Minisatellite maps give access to the detail of mutation processes at work on such loci. In this paper, we design an algorithm to compare two maps under an evolutionary model that includes deletion, insertion, mutation, tandem duplication and tandem deletion of a variant. Our method computes an optimal alignment in reasonable time; and the alignment score, i.e., the weighted sum of its elementary operations, is a distance metric between maps. The main difficulty is that the optimal sequence of operations depends on the order in which they are applied to the map. Taking the maps of the minisatellite MSY1 of 609 men, we computed all pairwise distances and reconstruct an evolutionary tree of these individuals. MSY1 (DYF155S1) is a hypervariable locus on the Y chromosome. In our tree, the populations of some haplogroups are monophyletic, showing that one can decipher a micro-evolutionary signal using minisatellite maps comparison.
Combinatorics of Periods in Strings
We consider the set Γ(n) of all period sets of strings of length n over a finite alphabet. We show that there is redundancy in period sets and introduce the notion of an irreducible period set. We prove that Γ(n) is a lattice under set inclusion and does not satisfy the Jordan- Dedekind condition.We propose the first enumeration algorithm for Γ(n) and improve upon the previously known asymptotic lower bounds on the cardinality of Γ(n). Finally, we provide a new recurrence to compute the number of strings sharing a given period set.
GeneNest: automated generation and visualization of gene indices
GeneNest is a collection of gene indices for several model species including mouse, Human, zebra fish and others. Each gene indice intends to gather all mRNA sequences available for one gene in a single group. This computation is performed by comparison and clustering of all available mRNA sequences including Expressed Sequence Tags (ESTs). GeneNest is accessible through a dedicated and powerful webserver. The all against all comparison is performed using QUASAR algorithm.
Exact and Efficient Computation of the Expected Number of Missing and Common Words in Random Texts
The number of missing words (NMW) of length q in a text, and the number of common words (NCW) of two texts are useful text statistics. Knowing the distribution of the NMW in a random text is essential for the construction of so-called monkey tests for pseudorandom number generators. Knowledge of the distribution of the NCW of two independent random texts is useful for the average case analysis of a family of fast pattern matching algorithms, namely those which use a technique called q-gram filtration. Despite these important applications, we are not aware of any exact studies of these text statistics. We propose an efficient method to compute their expected values exactly. The difficulty of the computation lies in the strong dependence of successive words, as they overlap by (q - 1) characters. Our method is based on the enumeration of all string autocorrelations of length q, i.e., of the ways a word of length q can overlap itself. For this, we present the first efficient algorithm. Furthermore, by assuming the words are independent, we obtain very simple approximation formulas, which are shown to be surprisingly good when compared to the exact values.
Transformation distances: a family of dissimilarity measures based on movements of segments
MOTIVATION: Evolution acts in several ways on DNA: either by mutating a base, or by inserting, deleting or copying a segment of the sequence (Ruddle, 1997; Russell, 1994; Li and Grauer, 1991). Classical alignment methods deal with point mutations (Waterman, 1995), genome-level mutations are studied using genome rearrangement distances (Bafna and Pevzner, 1993, 1995; Kececioglu and Sankoff, 1994; Kececioglu and Ravi, 1995). The latter distances generally operate, not on the sequences, but on an ordered list of genes. To our knowledge, no measure of distance attempts to compare sequences using a general set of segment-based operations. RESULTS: Here we define a new family of distances, called transformation distances, which quantify the dissimilarity between two sequences in terms of segment-based events. We focus on the case where segment-copy, -reverse-copy and -insertion are allowed in our set of operations. Those events are weighted by their description length, but other sets of weights are possible when biological information is available. The transformation distance from sequence S to sequence T is then the Minimum Description Length among all possible scripts that build T knowing S with segment-based operations. The underlying idea is related to Kolmogorov complexity theory. We present an algorithm which, given two sequences S and T, computes exactly and efficiently the transformation distance from S to T. Unlike alignment methods, the method we propose does not necessarily respect the order of the residues within the compared sequences and is therefore able to account for duplications and translocations that cannot be properly described by sequence alignment. A biological application on Tnt1 tobacco retrotransposon is presented. AVAILABILITY: The algorithm and the graphical interface can be downloaded at r̆lhttp://www.lifl.fr/ approximately varre/TD
q-gram based database searching using a suffix array (QUASAR)
With the increasing amount of DNA sequence information deposited in our databases searching for similarity to a query sequence has become a basic operation in molecular biology But even today s fast algorithms reach their limits when applied to all versus all comparisons of large databases Here we present a new data base searching algorithm dubbed QUASAR Q gram Alignment based on Su x ARrays which was designed to quickly detect se quences with strong similarity to the query in a context where many searches are conducted on one database Our algorithm applies a modi cation of q tuple ltering implemented on top of a su x array Two versions were de veloped one for a RAM resident su x array and one for access to the su x array on disk We compared our implementation with BLAST and found that our approach is an order of magnitude faster It is however restricted to the search for strongly similar DNA sequences as is typically required e g in the context of clustering expressed sequence tags ESTs.
Location of Repetitive Regions in Sequences By Optimizing A Compression Method
Suppose that a biologist wishes to study some local property P of genetic sequences. If he can design (with a computer scientist) an algorithm C which efficiently compresses parts of the sequence which satisfy P , then our algorithm TurboOptLift locates very quickly where property P occurs by chance on a sequence, and where it occurs as a result of a signi cant process. Under some conditions, the time complexity of TurboOptLift is O (n log n). We illustrate its use on the practical problem of locating approximate tandem repeats in DNA sequences.
Location of repetitive regions in sequences by optimizing a compression method
Suppose that a biologist wishes to study some local property $P$ of genetic sequences. If he can design (with a computer scientist) an algorithm $C$ which efficiently compresses parts of the sequence which satisfy $P$, then our algorithm TurboOptLift locates very quickly where property $P$ occurs by chance on a sequence, and where it occurs as a result of a significant process. The time complexity of TurboOptLift is $O(nłog n)$. We illustrate its use on the practical problem of locating approximate tandem repeats in DNA sequences.
Optimal representation in average using Kolmogorov complexity
One knows from the Algorithmic Complexity Theory [2-5, 8, 14] that a word is incompressible on average. For words of pattern $x^m$, it is natural to believe that providing $x$ and $m$ is an optimal average representation. On the contrary, for words like $x ⊕ y$ (i.e., the bit to bit or between $x$ and $y$), providing $x$ and $y$ is not an optimal description on average. In this work, we sketch a theory of average optimal representation that formalizes natural ideas and operates where intuition does not suffice. First, we formulate a definition of K-optimality on average for a pattern, then demonstrate results that corroborate intuitive ideas, and give worthy insights into the best compression in more complex cases.
Computational approaches to identify leucine zippers
The leucine zipper is a dimerization domain occurring mostly in regulatory and thus in many oncogenic proteins. The leucine repeat in the sequence has been traditionally used for identification, however with poor reliability. The coiled coil structure of a leucine zipper is required for dimerization and can be predicted with reasonable accuracy by existing algorithms. We exploit this fact for identification of leucine zippers from sequence alone. We present a program, 2ZIP, which combines a standard coiled coil prediction algorithm with an approximate search for the characteristic leucine repeat. No further information from homologues is required for prediction. This approach improves significantly over existing methods, especially in that the coiled coil prediction turns out to be highly informative and avoids large numbers of false positives. Many problems in predicting zippers or assessing prediction results stem from wrong sequence annotations in the database.
Detection of significant patterns by compression algorithms: the case of approximate tandem repeats in DNA sequences
Compression algorithms can be used to analyse genetic sequences. A compression algorithm tests a given property on the sequence and uses it to encode the sequence: if the property is true, it reveals some structure of the sequence which can be described briefly, this yields a description of the sequence which is shorter than the sequence of nucleotides given in extenso. The more a sequence is compressed by the algorithm, the more significant is the property for that sequence.We present a compression algorithm that tests the presence of a particular type of dosDNA (defined ordered sequence-DNA): approximate tandem repeats of small motifs (i.e. of lengths <4). This algorithm has been experimented with on four yeast chromosomes. The presence of approximate tandem repeats seems to be a uniform structural property of yeast chromosomes.The algorithms in C are available on the World Wide Web (URL: http://www.lifl.fr/~rivals/Doc/RTA/).E-mail: rivals@lifl.fr
Fast Discerning Repeats in DNA Sequences with a Compression Algorithm
Long direct repeats in genomes arise from molecular duplication mechanisms like retrotransposition, copy of genes, exon shuffling, … Their study in a given sequence reveals its internal repeat structure as well as part of its evolutionary history. Moreover, detailed knowledge about the mechanisms can be gained from a systematic investigation of repeats. The problem of finding such repeats is viewed as an NP-complete problem of the optimal compression of a sequence thanks to the encoding of its exact repeats. The repeats chosen for compression must not overlap each other as do the repeats which result from molecular duplications. We present a new heuristic algorithm, Search_Repeats, where the selection of exact repeats is guided by two biologically sound criteria: their length and the absence of overlap between those repeats. Search_Repeats detects approximate repeats, as clusters of exact sub-repeats, and points out large insertions/deletions in them. Search_Repeats takes only 3 seconds of CPU time for the genome of Haemophilus influenzae on a Sun Ultrasparc workstation.
A Guaranteed Compression Scheme for Repetitive DNA Sequences
We present a text compression scheme dedicated to DNA sequences. The exponential growing of the number of sequences creates a real need for analyzing tools. A specific need emerges for methods that perform sequences classification upon various criteria, one of which is the sequence repetitiveness. A good lossless compression scheme is able to distinguish between “random” and “significative” repeats. Theoretical bases for this statement are found in Kolmogorov complexity theory., keywords = DNA, sequences, dictionaries, encoding, complexity theory, tree data structures , sufficient conditions, biological information theory, Kolmogorov complexity
Compression and genetic sequence analysis
A novel approach to genetic sequence analysis is presented. This approach, based on compression of algorithms, has been launched simultaneously by Grumbach and Tahi, Milosavljevic and Rivals. To reduce the description of an object, a compression algorithm replaces some regularities in the description by special codes. Thus a compression algorithm can be applied to a sequence in order to study the presence of those regularities all over the sequence. This paper explains this ability, gives examples of compression algorithms already developed and mentions their applications. Finally, the theoretical foundations of the approach are presented in an overview of the algorithmic theory of information.
A First Step Towards Chromosome Analysis by Compression Algorithms
In this paper, we use Kolmogorov complexity and compression algorithms to study DOS-DNA (DOS: defined ordered sequence). This approach gives quantitative and qualitative explanations of the regularities of apparently regular regions. We present the problem of the coding of approximate multiple tandem repeats in order to obtain compression. Then we describe an algorithm that allows to nd efficiently approximate multiple tandem repeats. Finally, we briefly describe some of our results. Area : DNA sequence processing.